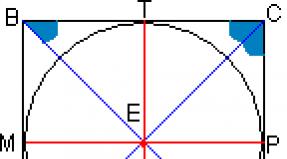
Bone marrow aplasia syndrome in newborns. Aplastic acquired anemia. Autologous bone marrow transplant
Artemyeva Veronika from Nizhny Tagil asks:
What is hypoplasia bone marrow, and what are the symptoms of this disease?
Expert answer:
Bone marrow hypoplasia is a condition in which myeloid tissue is replaced by adipose tissue. The very concept of "hypoplasia" in translation means the lack of formation. With insufficient formation of myeloid tissue, the function of the red bone marrow is disrupted, as a result of which the production of blood cells - leukocytes, erythrocytes, platelets - is significantly reduced. Bone marrow insufficiency is one of the varieties of pancytopenia.
Reasons for development
There are two forms of the disease:
- hereditary;
- acquired.
The following pathologies are the reason for the development of hereditary forms:
- Fanconi anemia;
- congenital dyskeratosis;
- Diamond-Blackfen anemia;
- other genetic diseases.
Insufficient production of blood cells can act as an independent disease in aplastic anemia or develop against the background of the following diseases:
- cirrhosis of the liver;
- chronic hepatitis;
- malignant neoplasms;
- various autoimmune disorders.
Disease manifestations
In the body of sick people, the volume of blood is much lower than that of healthy people. As a result of a decrease in the number of platelets, patients experience spontaneous bleeding. Any cuts, injuries that lead to significant blood loss can pose a danger. Mucous membranes and internal organs are prone to bleeding.
Insufficient production of leukocytes leads to a decrease in immunity, which contributes to the occurrence of frequent infectious diseases.
Treatment principles
This pathology is treated by a hematologist. The choice of the method of therapy depends on the cause of the disease. Aplastic anemia can be eliminated only by bone marrow transplantation. If it is not possible to find a suitable donor, the patient is shown taking drugs that inhibit immune system(Cyclosporin A). Immunosuppressive therapy can be successful only with mild forms of the disease.
All patients, without exception, receive intravenous administration platelet and erythrocyte mass. In order to prevent the development of infectious and fungal lesions, patients are prescribed antibacterial and antifungal drugs.
One of the reasons for the insufficient content of blood cells is an increase in the activity of the spleen - hypersplenism. Therefore, patients can undergo splenectomy - an operation during which the spleen is removed.
Video: What is Bone Marrow Transplant
© E.A. Orlova, S.V. Lashutin, 2004 UDC 616.419-003.978-02-08: 577.175.71
E.A. Orlova, S.V. Lashutin
TOTAL RED BONE MARROW APLASIA AS A RESULT OF ERYTHROPOETIN TREATMENT
E.A. Orlova, S.V. Lashutin
TOTAL APLASIA OF THE RED BONE MARROW AS A RESULT OF TREATMENT WITH ERYTHROPOIETIN
Department of Therapy and Occupational Diseases named after EAT. Tareeva Moskovskaya medical academy them. THEM. Sechenov, Russia
Key words: recombinant human erythropoietin, complete aplasia of the red bone marrow. Key words: recombinant human erythropoietin, pure red cell aplasia.
Recombinant human erythropoietin (rhEPO) immediately after registration in the late 1980s became the drug of choice in the treatment of anemia in patients with chronic renal failure (CRF). Side effects identified at the beginning of the use of the drug could be the result of a too rapid increase in hemoglobin (arterial hypertension, thrombosis, hyperkalemia) in combination with a direct effect on non-hematopoietic tissues (including vascular walls). Recently, complete aplasia of the red bone marrow (PAKKM) has become a serious problem, manifested by severe normocytic, normochromic anemia, a sharp decrease in the number of reticulocytes (< 10000/мм3), при нормальном количестве гранулоцитов и тромбоцитов и почти полном отсутствии эритроидных предшественников в пунктате костного мозга (менее 5% эритробластов, данные за блок созревания).
Due to the almost complete cessation of erythropoiesis, the hemoglobin concentration decreases very quickly, at a rate corresponding to the life span of erythrocytes (almost 0.1 g / dl / day, slightly less than 1 g / dl / week). Patients require weekly transfusions to maintain hemoglobin levels between 70 and 80 g / dL.
If from 1988, when rhEPO appeared on the market, to 1997, only 3 cases of PAKKM were registered, then in the last three years their number exceeded 100 (table). It should be noted that PAKKM was mostly associated with a single drug - eprex.
Etiology
PAKKM is a severe, aregenerative form of anemia, accompanied by aplasia of the bone marrow. Disease
is caused by epoetin-induced antibodies, which neutralize not only exogenous rhEPO, but also cross-react with endogenous erythropoietin. As a result, serum levels of erythropoietin cease to be determined, and erythropoiesis becomes ineffective.
Antierythropoietin antibodies after epoetin alfa therapy are polyclonal and are able to neutralize very high concentrations native EPO. These antibodies belong to class B, subclasses B1 or B4, and react with the protein portion of EPO. This has been demonstrated with the removal of carbohydrate residues by digestive enzymes without affecting the affinity of antibodies for erythropoietin. Thus, it is unlikely that glycosylation affects immunogenicity.
Epidemiology
The general population PACCM usually occurs spontaneously (in 50% of cases) or is associated with thymomas (in 5% of cases), lymphatic proliferative (myelodysplasia, B- and T-cell chronic lymphocytic leukemia and chronic myeloid leukemia) or immune (autoimmune hemolytic anemia, systemic lupus erythematosus, rheumatoid arthritis) diseases. It sometimes develops when certain medications are taken (anticonvulsants, antibiotics, and antithyroid drugs) or as a result of a viral infection (for example, parvovirus B19 or hepatitis B virus).
In adult patients, PAKKM is most often an autoimmune disease associated with the production and appearance of cytotoxic T-lymphocytes against erythropoietic progenitor cells or erythropoietic cells themselves. In rare cases, it is associated with the appearance of antibodies to endogenous erythropoietin in people who have never received rhEPO.
Cases of PACCM associated with antibodies to rhEPO in patients with chronic renal failure, according to the Pharmacological Research and Development Department of Johnson & Johnson
Eprex only 2 3 5 8 22 64 67 6 177
Other erythropoietins 1 0 1 0 3 5 5 3 18
Cases under investigation 5 2 0 5 11 16 18 6 63
Total number of suspected cases 8 5 6 13 36 85 90 15 258
Note. This implies the absence or decrease in the effect of rhEPO-therapy - an unexplained drop in hemoglobin levels or the need to increase the dose.
All published cases of RHECM associated with rhEPO relate exclusively to patients with chronic kidney disease (CKD), despite the widespread use of this drug in oncology. Cancer patients are probably less likely to develop this complication due to decreased immune status, other therapies, and shorter courses of epotherapy.
The first three cases of immuno-induced PACCM as a result of the use of rhEPO were detected between 1992-1997, and since 1998 there has been an increase in the prevalence of PACCM induced by antibodies to rhEPO.
Interestingly, the incidence of this complication per 10,000 patients per year was much higher for ep-rex (3.32) (data for the first half of 2002) than for epoetin-beta (0.12), epogen (0.02 ) and dar-bepoetin-alpha (0.5). In this regard, the Johnson & Johnson company issued a press release, which indicated that in 94.2% of cases of PAKKM after using eprex, the drug was injected subcutaneously. In December 2002, in the countries of the European Union, the annotation to eprex was amended: patients with chronic renal failure should receive the drug only intravenously. The measures taken led to a decrease in the incidence to 0.89 cases per 10,000 patients / year of admission by the first half of 2003. The instructions for the use of other erythropoietins did not change due to the lack of clear data that their use is associated with the risk of epoetin-induced PACCM. This, however, does not exclude that an increase in the incidence of AVCM as a result of subcutaneous injection other poets may be observed in the future.
The average age of the patients was 61 years, with some predominance of men. There was no correlation with the cause of renal failure.
sufficiency, treatment chronic illness kidney disease (CKD), age or gender, despite a disproportionately higher prevalence of this complication in men over 70 years of age, which predominates in the population of patients with end-stage renal failure (ESRD). Average duration erythropoietin treatment was 7 months prior to diagnosis of PACCM, ranging from 1 month to 5 years.
The structure of erythropoietin
There are currently three different types of rhEPO available on the market: epoetin-alpha, epoetin-beta, and epoetin-omega. All three molecules have the amino acid sequence of human epoetin, but differ in the number of polysaccharide chains and in the content of carbohydrates. Epoetin al-fa has slightly lower sialization than epoetin beta; this explains the small differences observed in the pharmacokinetics and pharmacodynamics of the two molecules, but this is unlikely to be the reason for their different immunogenicity.
Epoetin-omega contains lower amounts of O-linked sugar, is less acidic, and differs from the other two epoetins in hydrophilicity. Currently, there are no reports of PACCM cases in patients treated with epoetin-omega, but the population of patients treated with this drug is much smaller.
Darbepoetin alfa is a new product on the market. It contains five N-linked carbohydrate chains (two more than rhEPO), has a higher molecular weight, sialic acid content and a negative charge compared to other erythropoietins. Since the amino acid sequence and carbohydrate content of darbepoetin alpha is different from human EPO, it is theoretically possible that this new molecule could be immunogenic. But until now, the development of PAKKM with the use of this drug has not been observed.
Route of administration and other causes of immunogenicity
The rise in PACCM has coincided with a shift from intravenous to subcutaneous administration of rhEPO, especially outside the United States. It cannot be ruled out that the subcutaneous route of administration has a greater effect on immunogenicity than the intravenous route, because the skin has a highly developed immune system. It is possible that prolonged exposure of immunocompetent skin cells to epoetin after subcutaneous administration may increase immunogenicity. In addition, the subcutaneous route is associated with self-medication and increases the risk of inappropriate use or storage of the drug. The significance of storage conditions is not fully understood, but it is important that the drug is stored at temperatures between 2 ° and 8 ° C.
When conducting international studies, it was shown that the majority of patients with PACCM received the drug subcutaneously (94.2%). However, there are countries (for example, Italy) where PAACM was practically not detected, despite the fact that most patients received the drug subcutaneously.
The immunogenicity of rhEPO preparations can be influenced by factors that are not associated with differences between the endogenous and recombinant molecules. For example, the manufacturing process and ingredients that increase the potential for oxidation and aggregation, such as dry-freezing, can increase immunogenicity. The company ".Mochon & ICBN" came to the conclusion that the removal of human albumin from the composition of eprex in 1998, an increase in the frequency of subcutaneous administration (especially self-administration) and non-observance of storage conditions play a leading role in the development of PACCM with the use of eprex. The role of replacing human albumin with polysorbitol 80 (0.03% concentration) and glycine for stabilizing the composition of eprex is also not excluded. In epoetin-beta (neorecormone), polysorbitol-80 has been used as a stabilizer since the registration of the drug. In dar-beropoietin-alfa (aranesp), polysorbitol-80 is also used as a stabilizer (in lower concentrations - 0.005%), but cases of PAACM are not observed. As possible reasons The use of silicone oil as a lubricant for syringes has also been discussed since 1994. Most recent research focuses on organic compounds leached from the rubber plunger of eprex syringes with polysorbitol-80 solvent. The company says that they
have already replaced rubber pistons with Teflon coated pistons.
Diagnostics
Anti-rhEPO-induced PACCM is a serious but fortunately rare complication associated with epoetin treatment. The problem is being intensively studied by the authorities, manufacturers of erythropoietins, independent scientists, nephrological societies, but still remains unresolved.
Despite the rarity of PAKKM secondary to the treatment of rhEPO, doctors should be aware of this formidable complication and consider it in differential diagnosis in patients with rapidly increasing anemia and / or treatment resistance. The first step should be a complete examination to clarify the nature of the anemia (including assessment of the number of reticulocytes), exclusion of other known causes of anemia (iron deficiency, blood loss, infection, inflammation). The next step is bone marrow examination.
If PAKKM is detected, erythropoietin should be canceled immediately, anti-erythropoietin antibodies should be determined. Determination of antibodies is a key point in the diagnosis of AVCM. There is currently no standard screening method for detecting antibodies to epoetins. Available assays use either binding reactions or biological samples. Biological tests remain the only method that can reveal the neutralizing ability of antibodies. Other assays include radioimmune precipitation (RIP), used by N. Casadevall et al., And ELISA. Although no direct comparisons of methods have been published, RIP appears to be more reliable, while ELISA may have lower sensitivity and specificity. Although Amgen, Ortho Biotech, and Roche have all offered their anti-epoetin antibody test systems, independent laboratory test systems are preferred. Screening tests for antibodies to erythropoietin are recommended only for research studies. In the usual clinical practice in patients resistant to rhEPO therapy, in the absence of signs of PACCM in the bone marrow aspirate, there is no need to determine antibodies to erythropoietin.
Due to the fact that antibodies to hREPO are neutralizing and will cross-react both with all currently available exogenous erythropoietins and with endogenous erythropoietin, any erythropoietic
Kaya therapy should be discontinued immediately if PACCM is suspected.
Experience with PACCM treatment remains minimal. Nearly half of patients respond to immunosuppressants. The use of both corticosteroids and in combination with cyclosporine or cyclophosphamide, immunoglobulin or plasmapheresis has been described. Good results have been observed with steroids in combination with cyclophosphane, as well as with cyclosporine. The best results were observed in patients after kidney transplantation, probably because the immunosuppressive therapy prescribed after transplantation can be effective in AVCM.
After discontinuation of rhEPO, the antibody titer decreased slowly in all patients. It is assumed that immunosuppressants accelerated the decrease in antibody titer and, possibly, allowed to restore erythropoiesis to the level preceding erythropoietin therapy. Nevertheless, preliminary data show that almost 40% of patients remain dependent on blood transfusions even after 2 years of immunosuppressive therapy.
CONCLUSION
RhEPO therapy is a widely used treatment for renal anemia. This product of molecular genetic technology has been used for over 15 years and has an excellent therapeutic index (selective and powerful effect on erythropoiesis, accompanied by such
mi side effects as aggravating arterial hypertension or thrombotic complications). In pre-dialysis CKD patients, rhEPO also reduces morbidity and mortality, and also has a positive effect on cardiac function. In addition, the correction of anemia significantly improves the well-being and quality of life of patients. A noticeable increase in the prevalence of AVCM observed in last years, deserves special attention; however, we must balance its severity and extreme rarity with the high number of CKD patients who die each year from cardiovascular complications, which could be partially alleviated by treatment for anemia.
BIBLIOGRAPHIC LIST
1. Eckardt K-U, Casadevall N. Pure red-cell aplasia due to anti-erythropoietin antibodies. Nephrol Dial Transplant 2003 18: 865-869
2. Casadevall N, Nataf J, Viron B et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med 2002; 346: 469-475
3. Casadevall N, Dupuy E, Molho-Sabatier P et al. Autoantibodies against erythropoietin in a patient with pure red-cell aplasia. N Engl J Med 1996; 334: 630-633
4. Casadevall N. Antibodies against rHuEpo: native and recombinant. Nephrol Dial Transplant 2002; 17: 42-47
5. Locatelli F, Del Vecchio L. Pure red cell aplasia secondary to treatment with erythropoietin. Artificial Organs 2003; 27 (9): 755-758
6. Locatelli F, Aljama P, Barany P et al. Erythropoiesis-stimulating agents and antibody-mediated pure red-cell aplasia: where are we now and where do we go from here? Nephrol Dial Transplant 2004 19: 288-293
BONE MARROW APPLASIA
(a. medullae ossium) see Panmieloftiz.
Medical terms. 2012
See also the interpretations, synonyms, meanings of the word and what is BONE MARROW APLASIA in Russian in dictionaries, encyclopedias and reference books:
- APLASIA in the Popular Medical Encyclopedia:
- a developmental anomaly, in which a part of the body, organ or area of some ... - APLASIA in Medical terms:
(aplasia; a- + Greek plasis formation, formation; syn. agenesis) the general name of developmental abnormalities in which a part of the body, organ or ... - APLASIA in the Big Encyclopedic Dictionary:
(from a - negative prefix and Greek plasis - education) developmental defect, congenital absence of any part of the body or organ. Aplasia ... - APLASIA in big Soviet encyclopedia, TSB:
(from the Greek. a - negative particle and plasis - formation), agenesis, congenital absence of any part of the body or organ. A. arises ... - APLASIA
Congenital absence of any part of the body; the same as ... - APLASIA in the Encyclopedic Dictionary:
and, pl. no, well. Congenital absence of any part of the body; the same as ... - BRAIN
MOZGA INSTITUTE, organized in 1928, since 1954 in the system of the USSR Academy of Medical Sciences in Moscow. Since 1981 - Research Institute of the Brain as part of ... - APLASIA in the Big Russian Encyclopedic Dictionary:
APLASIA (from a - negative prefix and Greek plasis - education), developmental defect, congenital absence of K.-L. parts of the body or organ. ... - APLASIA in the Complete Accentuated Paradigm by Zaliznyak:
apla zia, apla zia, apla zia, apla ziy, apla zia, apla zia, apla zia, apla zia, apla zia, apla zia, apla zia, apla zia, ... - APLASIA in the New Dictionary of Foreign Words:
(a ... gr. plasis formation, formation) otherwise agenesis is a congenital absence of some kind. parts of the body due to disruption of the process of laying and development of tissues ... - APLASIA in the Dictionary of Foreign Expressions:
[a ... + gr. plasis formation, formation] otherwise agenesis - congenital absence of any. parts of the body due to a violation of the process of laying and development ... - APLASIA in the dictionary of Synonyms of the Russian language.
- APLASIA in the Dictionary of the Russian language Lopatin:
appl`az`ia, ... - APLASIA in the Complete Spelling Dictionary of the Russian Language:
aplasia, ... - APLASIA in the Spelling Dictionary:
appl`az`ia, ... - APLASIA in the Modern Explanatory Dictionary, TSB:
(from a - negative prefix and Greek plasis - education), developmental defect, congenital absence of any part of the body or organ. Aplasia ... - SPINAL CORD INJURY in the Medical Dictionary:
- BRAIN FAILURE in the Medical Dictionary:
Brain contusion - TBI, characterized by focal macrostructural damage to the medulla varying degrees severity. Diagnose in cases where cerebral ... - THROMBOCYTOPENIA in the Medical Dictionary:
- Immunodeficiency in the Medical Dictionary:
Immunodeficiencies are independent diseases (nosological forms) and concomitant syndromes characterized by a deficiency of the immune system. Frequency - 1 in 500 babies are born ... - Cerebral edema in the Medical Dictionary:
- SPINAL CORD TUMORS in the Medical Dictionary:
- BRAIN TUMORS in the Medical Dictionary:
- CHRONIC MYELOLEUKOSIS in the Medical Dictionary:
- ANEMIA APLASTIC in the Medical Dictionary:
- SPINAL CORD INJURY
Spinal cord injury is a variant of spinal cord injury characterized by the occurrence of spinal cord reversible (functional), as well as irreversible (organic) changes in ... - THROMBOCYTOPENIA in the Medical Dictionary:
Thrombocytopenia - reduced content platelets in peripheral blood, the most common cause of bleeding. With a decrease in the number of platelets less than 100x109 / l, the time is lengthened ... - Cerebral edema in the Medical Dictionary:
Cerebral edema (BEM) is an excessive accumulation of fluid in the brain tissue, clinically manifested by a syndrome of increased ICP; not a nosological unit, but ... - SPINAL CORD TUMORS in the Medical Dictionary:
Spinal cord tumors - tumors that develop from the parenchyma of the spinal cord, its roots, membranes or vertebrae; subdivided into extra- and subdural ... - BRAIN TUMORS in the Medical Dictionary:
Brain tumors - tumors that develop from the substance of the brain, its roots, membranes, as well as metastatic origin. Frequency. Tumors of the head ... - ANEMIA APLASTIC in the Medical Dictionary:
Aplastic anemias are a group of pathological conditions characterized by pancytopenia in the peripheral blood due to inhibition of the hematopoietic function of the bone marrow. Classification - Congenital ... - BONE MARROW HYPOPLASIA in Medical terms:
(hypoplasia medullae ossium) a bone marrow condition characterized by the replacement of the myeloid tissue of the bone marrow with adipose tissue and, as a result, a progressive decrease in intensity ... - HUMAN BRAIN: BRAIN RESEARCH in Collier's Dictionary:
Back to article HUMAN BRAIN Research on the brain is difficult for two main reasons. First, a direct line to the brain, reliably protected by the skull, is impossible ... - BONE in Encyclopedia Biology:
, the main element of the skeleton of vertebrates and humans. Bones serve as a receptacle for bone marrow and a depot of mineral substances in the body. Bone ... - MEAT PATE in the Book of Delicious and Healthy Food:
The meat canning industry produces canned meat pates in the following assortment: "Liver", "Moskovsky", "Arctic", "Diet", "Diet with brains", "Myasnoy" pate. Part … - CRANIAL INJURY in the Medical Dictionary:
Craniocerebral trauma Traumatic brain injury (TBI) - mechanical energy damage to the skull and intracranial contents ( meninges, vessels, cranial nerves). ... - SPINAL SPINAL INJURY in the Medical Dictionary:
Spinal cord injury - damage by mechanical energy to the spine and spinal cord. Causes changes both in the spinal cord itself and in ... - PARANEOPLASTIC SYNDROME in the Medical Dictionary:
Many carcinomas are accompanied by the development of characteristic symptoms that are not directly determined by the tumor itself - paraneoplastic syndromes. These include endocrine, hematological syndromes ... - WARDENBURG'S SYNDROME in the Medical Dictionary.
- THALASSEMIES in the Medical Dictionary:
Thalassemia is a hypochromic microcytic anemia with inherited abnormalities in globin genes. Thalassemya is common in the Mediterranean countries, the Middle East and ... - Cranial Facial Dysplasia in the Medical Dictionary:
Craniofacial dysplasias are an extensive group of developmental defects with pronounced deviations in the shape of the facial skeleton, often with hypertelorism. The gene involved is usually ... - OSTEOMYELITIS in the Medical Dictionary:
- MULTIPLE MYELOMA in the Medical Dictionary:
- PREGNANCY in the Medical Dictionary:
Prematurity is a condition of a fetus born before the end of the normal period of intrauterine development (before the expiration of 37 weeks of gestation), with a body weight of less ... - DENDIWALKER MALFORMATION in the Medical Dictionary:
DANDY-WALKER MALFORMATION Dandy-Walker malformation is an anomaly in the development of the IV ventricle in combination with cerebellar hypoplasia, hydrocephalus, atresia of Lyushka's orifices and ... - Atresia of the posterior passage and rectum in the Medical Dictionary.
- LYMPHOMA NEDHODJKENSKAYA in the Medical Dictionary:
- POLICYTHEMIA TRUE in the Medical Dictionary:
- RADIATION DISEASE in the Medical Dictionary.
- CHROMOSOMAL DISEASES in the Medical Dictionary:
Chromosomal diseases are a large group of diseases (more than 300 syndromes) caused by abnormalities in the number or structure of chromosomes. Pathological changes in chromosomal ... - ANEMIA in the Medical Dictionary:
Anemia is a decrease in the amount of Hb in a unit of blood volume, more often with a simultaneous decrease in the number of red blood cells (or the total volume of red blood cells). The term ... - ALEYKIA ALIMENTARYNOTOXIC in the Medical Dictionary:
ALEIKIA ALIMENTARY-TOXIC Alimentary-toxic aleukia is food mycotoxicosis that occurs when eating products from crops that have wintered in the field (millet, buckwheat, wheat, ... - ACUTE LEUKEMIA in the Medical Dictionary:
Acute leukemia - malignant disease hematopoietic system; morphological substrate - power cells. Frequency. 13.2 cases per 100,000 population among men ... - HYDRONEPHROSIS in the Medical Dictionary:
Hydronephrosis is a persistent and progressively increasing expansion of the pelvis and calyces (normal capacity 3-10 ml). Frequency. Prevalence among children - 2% ... - Hypoparathyroidism in the Medical Dictionary:
Hypoparathyroidism is a syndrome of insufficient function of the parathyroid glands, characterized by seizures, nervous and mental disorders, a decrease in the content of calcium in the blood. - Postoperative ... - POLICYTHEMIA TRUE in the Medical Dictionary:
Polycythemia vera (polycythemia vera) is a neoplastic disease, accompanied by an increase in the number of red blood cells, white blood cells and platelets. The source of tumor growth is the myelopoiesis precursor cell. ... - OSTEOMYELITIS in the Medical Dictionary:
Osteomyelitis is an acute or chronic inflammation of the bone marrow, usually involving the compact and cancellous bone and periosteum. Prevailing age ... - MULTIPLE MYELOMA in the Medical Dictionary:
Multiple myeloma - a tumor from plasma cells (differentiated B-lymphocytes), secreting monoclonal pathological Ig (paraprotein). The disease belongs to the group of paraproteinemic hemoblastoses. ... - CHRONIC MYELOLEUKOSIS in the Medical Dictionary:
Chronic myeloid leukemia (CML) is characterized by the proliferation of cells of monocytic and granulocytic origin with an increase in the number of leukocytes in the peripheral blood up to 50x109 / l higher. ... - LYMPHOMA NEDHODJKENSKAYA in the Medical Dictionary:
Non-Hodgken lymphoma is a heterogeneous group of diseases characterized by neoplastic proliferation of immature lymphoid cells that accumulate outside the bone marrow. Lymphosarcomatosis (Kundrat's disease) - ...
Many carcinomas are accompanied by the development of characteristic symptoms that are not directly determined by the tumor itself - paraneoplastic syndromes. These include endocrine, hematological syndromes ...
Aplastic anemia is a severe hematological disease, accompanied by anemia, a sharp decrease in immunity, as well as disorders of blood coagulation processes. It occurs due to suppression of the hematopoietic function of the bone marrow (or bone marrow aplasia).
The disease was first described by the famous German physician and scientist Paul Ehrlich, in 1888. A previously unknown pathology found in a young pregnant woman was accompanied by severe anemia, a decrease in the number of leukocytes, fever, bleeding, and quickly led to the death of the patient. The postmortem examination revealed the replacement of the red bone marrow with adipose tissue. Later, in 1907, Anatole Chaoffard, a French physician, suggested calling this disease aplastic anemia.
Aplastic anemia is a fairly rare disease. The average incidence is 3-5 per 1 million of the total population per year. Most of the patients are children and young people.
Types of aplastic anemias
Distinguish between hereditary (genetically determined) and acquired aplastic anemia.
80% of cases of the disease are due to the acquired form of pathology, 20% are caused by genetic factors.
Doctors use the ICD-10 classification of pathology ( International Classification Diseases of the 10th revision). There are the following types of aplastic anemias:
D61.0 Constitutional aplastic anemia
D61.1 Drug-induced aplastic anemia
D61.2 Aplastic anemia due to other external agents
D61.3 Idiopathic aplastic anemia
D61.8 Other specified aplastic anemias
D61.9 Aplastic anemia, unspecified
Aplastic anemia in children
In children, in most cases, the disease is acquired in nature. The incidence is 2-3 cases per 1 million children (the peak incidence occurs in adolescence). In 70% of cases, the direct cause of the disease cannot be established, it is generally accepted that viral infections, chemicals and medications.
Most often, the diagnosis is made by chance, when general analysis blood. At correct treatment and timely diagnosis the prognosis is favorable. Aplastic anemia in children is well treated. The results of using bone marrow transplantation and immunosuppressive therapy are approximately the same in terms of effectiveness, however, preference should be given to bone marrow transplantation from a suitable (ideally a brother or sister) donor. Modern methods treatment of aplastic anemia in childhood allow you to maintain health and do not affect the ability to have children in the future.
Causes and risk factors for aplastic anemia
Genetically determined disorders of hematopoietic function are noted in some hereditary pathologies such as familial Fanconi anemia, Schwachman-Diamond syndrome, true erythrocyte aplasia, congenital dyskeratosis.
Mutations in critical genes that regulate the cell cycle, protein synthesis, and protect and repair DNA damage lead to the formation of defective stem (hematopoietic) cells. Errors in the genetic code trigger apoptosis, the mechanism of programmed cell death. At the same time, the stem cell pool shrinks much faster than in healthy people.
The acquired form of pathology occurs as a result of direct toxic effects on hematopoietic cells. These factors include:
· Exposure to ionizing radiation. Maria Sklodowska-Curie, physicist, twice Nobel Prize winner, received for her work in the field of radioactivity research and for the discovery of new radioactive elements, died from aplastic anemia;
· Pesticides, insecticides, benzene derivatives, salts of heavy metals, arsenic have a direct toxic effect on the bone marrow, inhibit the production of blood corpuscles and lead to the death of stem cells;
· Some medications have a similar effect. Non-steroidal anti-inflammatory drugs, antineoplastic drugs, analgin, chloramphenicol (causes the most severe form of the disease, which, according to statistics, occurs in 1 out of 30 thousand courses of treatment with chloramphenicol), mercazolil, carbamazepine, quinine can cause aplastic anemia in some people;
· Viruses can be a triggering factor for disease. Viral hepatitis, some types of parvoviruses, CMV, Epstein-Barr virus and HIV have the ability to cause a malfunction in the immune system, as a result of which it begins to attack own fabrics organism. For example, 2% of patients with acute viral hepatitis- reveal aplastic anemia;
· Autoimmune diseases(rheumatoid arthritis, SLE) may also be accompanied by bone marrow aplasia;
· Aplastic anemia in pregnancy is also thought to be due to a disorder in the immune system.
In more than 50% of cases, the immediate cause of the disease is not found, then they talk about idiopathic aplastic anemia.
What Happens With Aplastic Anemia
Red bone marrow is the main and most important hematopoietic organ in which the formation and maturation of blood elements occurs. Hematopoietic stem cells in it give rise to erythrocytes (responsible for the transfer of O 2 and CO 2 ), leukocytes (provide immunity) and platelets (participate in blood clotting processes). The number of hematopoietic cells is limited and gradually decreases throughout a person's life.
With aplastic anemia, there is a massive death of bone marrow stem cells, and, as a result, a sharp decrease in the content of erythrocytes, platelets and leukocytes in the patient's bloodstream. A lack of red blood cells leads to anemia, a decrease in the number of leukocytes causes a sharp suppression of the immune system, a decrease in the number of platelets is the cause of bleeding and, as a result, an increased risk of uncontrolled bleeding.
The results of recent studies suggest that the acquired form of the disease is almost always an autoimmune pathology. The key moment in the development of red bone marrow aplasia is the direct cytotoxic effect of T-lymphocytes. However, the reason why T-lymphocytes begin to recognize hematopoietic stem cells as targets for attack is still unknown. Point mutations in genes encoding human leukocyte antigens (HLA system) and explaining the distorted immune response (as in other autoimmune pathologies) can serve as a trigger factor.
It is also believed that for the development of pathology, a combination of several factors is required - both internal (unknown defects in stem cell DNA, HLA gene mutations, immune disorders) and external (drugs, viral infections, exotoxins and antigens).
How to suspect aplastic anemia - symptoms and signs of the disease
Symptoms characteristic of the disease:
· Unexplained weakness, fatigue, drowsiness;
· Low efficiency;
Shortness of breath that occurs even with mild physical exertion;
Dizziness, headaches;
Interruptions in the heart, palpitations, tachycardia;
· Pallor of the skin;
Prolongation of blood clotting time, hemorrhage in soft tissue, brain, bruising and bruising with minor exposure, nosebleeds, prolonged debilitating menstruation in women;
· Small-point hemorrhages in the skin and mucous membranes, bleeding of the gums;
· Frequent infections (respiratory tract, skin, mucous membranes, urinary tract), accompanied by fever;
· Painless ulcers on the oral mucosa;
· Weight loss, weight loss.
The course of the disease can be gradual or fulminant (with rapid development extremely severe anemia, immunodeficiency, disorders of blood coagulation processes with corresponding complications).
Diagnosis of aplastic anemia
For diagnostics, a detailed blood test and a histological examination of the material obtained from the bone marrow are used.
Laboratory signs of pathology found in peripheral blood:
· Decrease in the concentration of erythrocytes and hemoglobin in the blood without iron deficiency;
· Decrease in the concentration of all types of leukocytes in the patient's blood;
· Deficiency of platelets;
· Low number of reticulocytes - immature forms of erythrocytes;
· Increase in erythrocyte sedimentation rate (up to 40-60 mm / h).
In very severe cases, the hemoglobin concentration falls below 20-30 g / l. Color index, levels of serum iron, erythropoietin are usually normal or elevated. The platelet count is below normal, in severe cases they are completely absent.
The diagnosis is confirmed with a bone marrow biopsy. Punctate histology shows a high fat content on the background of a decrease in the number of hematopoietic cells. Cellularity (the total content of hematopoietic stem cells) is below 30%, megakaryocytes - platelet precursor cells may be absent.
Severity of aplastic anemia
Based on the results of a biopsy, aplastic anemia of mild, severe and extremely severe degrees is distinguished.
Severe form of the disease: cellularity - below 25%; in peripheral blood: neutrophils -< 0,5х10 9 / l, platelets -< 20х10 9 / l, reticulocytes -< 20х10 9 / l.
Extremely severe form of the disease: cellularity - below 25; in peripheral blood: neutrophils -< 0,2х10 9 / l, platelets -< 20х10 9 / l, reticulocytes -< 20х10 9 / l.
An easy form of pathology, deviations from the norm do not reach such critical indicators.
Aplastic anemia treatment
Treatment tactics depend on several factors: on the severity, age of the patient, the possibility of a bone marrow transplant from a suitable donor (ideally, close consanguineous relatives of the patient).
Bone marrow transplantation from a suitable donor is considered to be the optimal method for treating severe and extremely severe pathology. The maximum effect is observed in young patients. With a bone marrow transplant from a suitable donor, the 10-year survival rate can be as high as 85-90%.
If there are contraindications to bone marrow transplantation or if it is not possible to carry it out (no suitable donor), immunosuppressive therapy is used.
The main drugs used for conservative therapy are antithymocyte immunoglobulin (ATG) and cyclosporin A.
ATG is a serum containing antibodies against human T lymphocytes obtained from horse blood. The introduction leads to a reduction in the population of T-lymphocytes in the patient's body, as a result, the cytotoxic effect on stem cells decreases, and the hematopoietic function improves.
Cyclosporin A is a selective immunosuppressive agent that selectively blocks the activation of T-lymphocytes and the release of interleukins, including interleukin-2. As a result, the autoimmune process that destroys stem cells is blocked, and hematopoietic function improves. Cyclosporin A does not suppress the hematopoietic function of the bone marrow and does not lead to total immunosuppression.
Indications for the appointment of glucocorticosteroids in aplastic anemia are limited to the prevention of complications during ATG therapy. In all other cases, steroid hormones are of mediocre effectiveness and are the cause of a number of complications.
In spite of high efficiency immunosuppressive therapy, the most radical treatment is bone marrow transplant. The use of ATG and cyclosporin A increases the risk of developing myelodysplastic syndrome and leukemia, does not guarantee the absence of relapses of the disease.
If immunosuppressive therapy is ineffective, a bone marrow transplant is performed from a donor who is not related to the patient. The results of the surgery may vary. In 28-94% of cases, 5-year survival is noted, in 10-40% of cases, graft rejection occurs.
Patients with severe aplastic anemia receive blood products as an emergency medical care... Red blood cell transfusion allows you to quickly compensate for anemia, and platelet transfusion prevents life-threatening bleeding.
Correct lifestyle for aplastic anemia
Even with persistent remission, it is necessary to undergo periodic examinations (first of all, to take blood tests) and, if possible, avoid exposure to negative factors.
During the treatment period, it should be remembered that patients with aplastic anemia have a weak immune system. It is necessary to avoid visiting crowded places, wash your hands regularly, and do not eat food prepared in questionable places (due to the risk of infection). Timely vaccination can prevent some diseases (including the flu).
The high risk of bleeding or hemorrhage limits sports, especially those that are traumatic. Despite this, an active lifestyle with regular dosage physical activity positively affect the well-being and psycho-emotional state of patients.
A balanced diet rich in vitamins, minerals and proteins contributes to quick recovery hematopoiesis. Perishable foods should not be consumed (due to the risk of foodborne infections). When treating with cyclosporine A, salt intake should be limited.
Complications of aplastic anemia
Opportunistic infections (viral, fungal, bacterial) caused by immunodeficiency;
Bleeding, hemorrhage, blood clotting disorders (due to a low platelet count);
Complications due to side effects of drugs for the treatment of aplastic anemia (secondary hemochromatosis, serum sickness);
Transformation of the disease into myelodysplastic syndrome, leukemia and other hematological diseases.
Prognosis for aplastic anemia
Until the causes and mechanisms of the development of pathology were clarified, mortality from aplastic anemia reached 90%. Over the past 20-30 years, it has been possible to significantly reduce the mortality of the disease. Modern treatment methods have significantly improved the prognosis - 85% of patients reach the 5-year survival threshold.
In children and young people, with adequate treatment, the prognosis is favorable and the five-year survival rate reaches 90% (for patients over 40 years old - 75%).
Prevention of aplastic anemia
Currently, there are no effective measures for the prevention of genetically determined aplastic anemia.
Prevention of acquired aplastic anemia consists in adequate protection against exposure to toxic substances, pesticides and ionizing radiation. Self-administration of medicines should be avoided, especially long-term and in high doses.
Aplastic anemia is a failure of the hematopoietic system, which is expressed as normocytic normochromic anemia with concomitant thrombopenia and granulocytopenia. Intensity various symptoms is different and fluctuates in different periods of the disease. The name "aplastic anemia" is not accurate, because the pathological process selectively affects not only the red blood system, but all three main systems of the bone marrow. The name "panmyelophthisis" is also inaccurate, which speaks of atrophy of all cellular elements of the bone marrow, which is not always confirmed by biopsy. There are often cases of this disease in which the bone marrow is not "empty".
When studying the myelogram, a reduced, normal or even increased number of cells can be found, and young, immature elements, functionally deficient, may predominate. It should be remembered that the picture of the bone marrow from different parts the skeleton of the same patient may be different. Therefore, the concept of insufficiency of the entire bone marrow system best expresses the essence of the disease, since it simultaneously encompasses various manifestations of bone marrow aplasia, both morphological and functional.
Aplasia is not a definition of a nosological unit, but a complex of symptoms caused by various known and unknown pathogenetic factors. Recently, these syndromes are increasingly recognized in children, and the so-called idiopathic aplastic anemia, i.e. one in which the cause is not always known is rare in infancy.
Pathogenesis... Aplastic anemia can develop with various general diseases of the body, endogenous intoxication, with infections, as well as under the influence of toxic medicinal substances and chemical compounds used in industry and households, as well as under the influence physical factors(especially ionizing radiation). In addition, conditions in which the normal structure of this organ is replaced by tumor metastases or hyperplasia of fibrous tissue are among the more rare causes of bone marrow failure in infants and young children.
More and more importance in the pathogenesis of aplastic syndromes has recently been attributed to medicinal substances and chemical compounds, as well as to the effect of ionizing radiation. These factors can be divided into two groups, and in the first, as a rule, aplasia or hypoplasia of the bone marrow develops, if the dose of this factor is large enough or the time of its action is long enough. Another group includes those chemical compounds that cause the symptoms of aplasia only when they are accompanied by the child's individual hypersensitivity to this factor.
The factors that act depressively on the hematopoietic function of the bone marrow and can lead to its failure include, first of all, all types of radiation energy (X-rays, radioactive elements used in medicine), benzene, cytostatic and antimetabolic agents used in the treatment of leukemia and other proliferative processes, such as nitrogen mustard gas and its derivatives, nitrogranulogen, TEM, E39, nitromine, leukeran, milleran, urethane, aminopterin, ametopterin, 6-mercaptopurine and others.
Among the chemical compounds and drugs that cause symptoms of insufficiency of the entire structure of the bone marrow, organic compounds of arsenic (neoarsenphenamine), sulfonamides, antiepileptic drugs, gold preparations, athebrine, antihistamines and some antibiotics should be named. From this point of view, the toxic effect of chloromycetin on the bone marrow in children deserves special attention. Other antibiotics that occasionally cause bone marrow aplasia include streptomycin, and even penicillin and sometimes terramycin. Of the 334 children with aplastic anemia under the supervision of this author, 21.4% had a history of allergy symptoms. The percentage of allergies was even higher (62.4%) in the group of patients with aplastic syndrome after treatment with chloromycetin and other antibiotics.
Another group of substances that are potentially unsafe and can have a depressive and toxic effect on the hematopoietic system includes a number of chemical compounds used in the economy and industry. These include paints, varnishes, paint thinners, fluids used for cleaning metals, disinfestants such as DDT, azotox, dyes, as well as some cosmetics and others. Some of them, such as compounds containing benzene, enter the body through the respiratory tract and can imperceptibly, with longer exposure in poorly ventilated rooms, lead to bone marrow damage.
The above list of chemicals that are harmful to the hematopoietic system is not complete. Their number is growing all the time due to the increasingly widespread use in Everyday life substances of industrial origin.
Some aplastic symptom complexes can probably develop on the basis of autoimmune mechanisms. However, cases in which there are antibodies directed against all three bone marrow systems (pancytopenia) are rare. Immune syndromes are more common in which antibodies are directed selectively against elements of red blood, platelets or granulocytes. Aplastic anemia can develop when various diseases and general infections leading to inhibition of hematopoiesis). Pancytopenia has been described in disseminated tuberculosis, typhoid fever, general infection, and even in severe influenza and pneumonia. Cases of aplastic anemia due to focal infection and with rheumatism.
In newborns, the cause of bone marrow aplasia can be congenital syphilis, toxoplasmosis, and generalized megalocytosis caused by the virus of the salivary glands. except bacterial infection Infection with protozoa - chronic malaria, kala-azar and others - can lead to pancytopenia. In these syndromes, bone marrow suppression is attributed to an enlarged spleen. The same phenomenon can occur in pathological processes occurring in this organ (tuberculosis, sarcoidosis, and others). A large spleen with hyperplasia of the reticuloendothelial system can retain and destroy individual blood cells. In addition, this organ can have a long-term depressive effect on the structure of the bone marrow.
Another pathogenetic group consists of cases occurring in the form of aplastic anemia, in which the picture of aplasia is the initial stage of acute leukemia. In these cases, the course of the disease can be quite long (from several months to 1.5 years), the essence of the disease may remain unexplained until the last, acute phase, and sometimes it becomes clear only on the sectional table. In this case, it does not always come to a significant increase in the liver and spleen, in contrast to typical leukemia in children. Large differential diagnostic difficulties are presented by cases with bone marrow, poor in formed elements, with a predominance of small lymphocytes, often found in poor cell myelograms in aplastic syndromes. In these cases, it is difficult to decide whether these cells are true lymphocytes or micromyeloblasts.
In addition to cases of general insufficiency of the structure of the bone marrow in children due to known factors, a group of so-called idiopathic aplasia should be distinguished, in which all attempts to detect the causative factor are unsuccessful. In infancy, such conditions are rare; in the future, they are likely to occur less and less in view of the growing possibilities for more accurate detection of the pathogenetic factors of this symptom complex. It should be emphasized that both the clinical and hematological picture, in cases of idiopathic and symptomatic, is basically very similar. Therefore, it is difficult to draw a line between these phenomena.
From a theoretical point of view, the prognosis is better when the pathogenic factor is known and can be removed (eg medication). However, in almost every case, regardless of causative factor, the prognosis is very serious and the fate of patients is difficult to foresee.
Clinical picture aplastic anemia in children can be influenced by many conditions and factors. These primarily include the age of the child and general state his. Then the type and nature of the pathogenetic factor, dose and exposure, the degree of damage to individual elements of the bone marrow, the regenerative abilities of the hematopoietic system, the presence of concomitant infection, malnutrition, vitamin deficiency and much more play a role. Depending on the susceptibility of individual cellular systems of the bone marrow, they suffer to varying degrees, which determines the variability of the intensity of hematological and clinical symptoms.
The disease begins imperceptibly. Frequent and early symptoms include increasing general weakness, easy fatigue, lack of appetite, and pallor of the integument. Many children already at an early stage have thrombopenia with symptoms of hemorrhagic diathesis (nosebleeds, hemorrhages into the skin), while granulocytopenia may not manifest itself clinically for a long time. Anemia is normocytic and normochromic. Less commonly, there are single macrocytes in blood smears.
Sometimes celebrated lung symptoms hemolysis or the so-called symptom of a shortened life span of erythroblasts is determined. With the course of the disease, anemia and granulocytopenia progress. In some cases, the earliest hematological symptom is leukopenia, and this symptom can be long ahead of the manifestation of anemia and thrombopenia.
Peripheral blood picture predominantly monotonic, there is no pronounced aniso- and poikilocytosis, as well as erythrocyte polychromatophilia. In the qualitative composition of white blood, attention is drawn to the relative lymphocytosis. The number of reticulocytes is sharply reduced, except for those cases in which hemolytic mechanisms are attached.
The picture of the bone marrow in aplastic anemia is heterogeneous, from hypoplastic to normal cellular and even hyperplastic. In these latter cases, there is usually a clear shift to the left in the row of erythroblasts and granulocytes (functional bone marrow failure, delayed maturation). Patients with bone marrow, poor in cellular elements, usually have more or less lymphocytes and cells of the reticuloendothelial system. In addition, in the bone marrow, in the absence of erythroblasts, granulocytes and megakaryocytes, plasmocytes can be found, and sometimes tissue basocytes (mast cells), which do not exist in a normal myelogram. The variety of the bone marrow picture may depend not only on different pathogenesis, but also on the intensity of the pathogenetic factor. It is known that the same pathogenetic factor (for example, benzene or radiation energy) can cause damage to the bone marrow from complete aplasia to myelofibrosis with the formation of foci of extramedullary hematopoiesis, and even to conditions that cannot be distinguished from leukemia.
Bone marrow examination, regardless of the variety of changes in aplastic syndrome, is important. It allows you to assess the state of its individual systems and, to a certain extent, make a correct forecast. On the basis of the myelogram, other syndromes can be excluded. The greatest differential diagnostic difficulties arise in aplastic conditions and acute leukemia. It should be emphasized that a single study of the bone marrow is not always sufficient in these cases due to the evolution of the pathological process. It should also be remembered that the detection of "empty" bone marrow in a biopsy material from one bone does not prejudge the cellular composition of the bone marrow in other parts of the skeleton.
The course of aplastic anemia changes over time. As anemia, hemorrhagic diathesis and granulocytopenia increase, an exacerbation of the disease may occur, which initially proceeded chronically. The course of the disease may depend on a decrease in the general resistance of the body and the addition of a local or general infection. In this case, a febrile state develops and the disease enters the terminal phase - general sepsis develops. In most cases, aplastic syndrome in children proceeds slowly, gradually progressing, and the pathological symptoms gradually increase. A common reason death is a cerebral hemorrhage or bleeding from gastrointestinal tract... In some cases, the disease runs violently from the very beginning and quickly ends in death from sepsis. In rare cases with chronic course spontaneous remissions occur.
Based on the material from Wolf "a 334 children, 7.3% of children managed to survive for more than 5 years. More than half of the children died within a year from the moment the diagnosis was made; only 3% of children were cured.
Treatment chronic aplastic anemia consists primarily in the removal of the pathogenic factor, if known, and in the appointment symptomatic remedies... The latter include: blood transfusion, erythrocyte suspension, and, if necessary, platelet suspension transfusion. The role of transfused blood is primarily a substitutional one, since it was not possible to prove its stimulating effect on the function of the bone marrow or an increase in reticulocytosis.
Attempts to transfuse or transplant bone marrow seem to be theoretically justified, but so far the results of this treatment are inconclusive. Blood transfusion can usually provide temporary improvement, but over time, hemotherapy in the treatment of aplastic conditions becomes less effective due to the sensitization of patients to some antigenic factors of the transfused blood.
When treating aplastic syndromes, use hormonal drugs: ACTH, cortisone, prednisone, methylprednisolone, dexamethasone, etc. These drugs reduce the symptoms of hemorrhagic diathesis; sometimes they can be useful, protecting the body from possible post-transfusion reactions, often violent. They usually do not affect the blood picture and the general course of the disease.
If the course of the disease is complicated by an infection, the use of certain antibiotics in combination with vitamins (vitamin B complex, ascorbic acid) is indicated. In general, the treatment of aplastic anemia in children is the most difficult and unfavorable task.
Shahidi and Diamond observed remission with a regenerative response and reticulocytosis in several cases of bone marrow aplasia in children under the influence of testosterone treatment. After exhaustion of all therapeutic measures in the absence of improvement, in some cases they tried to resort to splenectomy. Of the 35 children who underwent this operation on the Wolf "a material, only one case was cured. Splenectomy can be justified in cases with bone marrow rich in cellular elements if there is indirect evidence that the spleen is an organ that is also responsible for anemia and pancytopenia.