Paroxysmal night hemoglobinuria (PNG). How does paroxysmal night hemoglobinuria manifest? What to do if positive APG tests
The reasons:
Causes of disease are associated with intravascular destruction of erythrocytes, defective in a significant part. Along with the pathological population of erythrocytes, some of the normal cells that have a normal life lifetime are also preserved. Disruptions in the structure of granulocytes and platelets are found. The disease is not hereditary, but any external factors provoking the formation of a defective population of cells, which is a clone, i.e. The offspring of one initially modified cell is not known.Thrombotic complications for APG are associated with intravascular hemolysis provoking thrombosis. The origin of an important, but far from the mandatory sign of the disease - paroxysis of hemoglobinuria at night or in the morning - remains unclear.
Paroxysism is not associated with the time of day, but with a dream, which may also cause crisis during the day. There is an increased complementaryness of pathological erythrocytes with APG. Perhaps this is the basis for provocating a hemolytic crisis with a transfusion of fresh blood, which contains factors that activate complement. The transfusion of blood stored for more than a week, hemolysis does not provoke.
Symptoms of paroxysmal night hemoglobinuria:
The disease develops slowly: signs of moderate anemia, weakness, fatigue, heartbeat under load, abdominal pain, often associated with thrombosis of mesenterial vessels appear. The skin and mucous membranes are pale yellow, grayish in connection with anemia and hemosideric deposition. Characteristic signs of intravascular hemolysis.The appearance of black urine is a non-permanent sign. Since the APG is often accompanied by leukopenia (mainly due to granulocyptopenia), chronic infectious complications are possible. Thrombocytopenia may complicate hemorrhagic syndrome. Long-term separation with urine hemoglobin and hemosiderin gradually leads to the development of iron deficiency - an asthenic syndrome occurs, dry skin, nail fragility appears.
The picture of the blood is characterized first normal, and then hypochromic anemia, small reticulocytosis (2-4% or more), leukopenia and thrombocytopenia. The morphology of erythrocytes does not have characteristic signs. In the bone marrow there is a hyperplasia of a red sprout, but in Trepanate there is a slight increase in the bone marrow cell, which, as the disease, it can become hypoplastic.
Due to the constantly current intravascular hemolysis in the plasma, the content of free hemoglobin was increased (normally less than 0.05 g / l). The level of serum iron is first normal, then can be significantly reduced. Along with the typical beginning of the disease, when hemolytic syndrome prevails, the picture of aplastic syndrome, which in a few years can be complicated by a hemolytic crisis with typical night hemoglobinuria. More often, hemolytic crisis provokes blood transfusion.
Diagnosis:
The diagnosis is established on the basis of signs of intravascular hemolysis (anemia, small reticulocyte, hemosiderer in the urine). Specify the diagnosis with special studies (positive sucrose sample, hem test, negative Cumbac sample).A gemolysin form of autoimmune hemolytic anemia, which is similar to external manifestations of Auhimmune hemolytic anemia, is characterized by the presence of hemolysins in serum, positive Kumbas breakdown. In contrast to the APG, there is no leukopenia and thrombocytopenia, usually a good effect gives prednisolone. DEVERATE FROM APLASTIC ARMIA PHB Allows the pattern of bone marrow: in Aplasia, the trepanat is characterized by the predominance of fat, during hemolysis - cell hyperplasia, but in rare CHANS, the pattern of bone marrow hypoplasia can develop, although hemosidemin is constantly detected in the urine, and reticulocytosis in the blood.
Treatment of paroxysmal night hemoglobinuria:
Treatment in the absence of pronounced anemia is not carried out. Heavy anemic syndrome requires transfusion of red blood cells; The best results give the transfusion of washed or weathered within 7-10 days of erythrocytes. In the heap formation hypoplace, anabolic steroids are shown: Nerochol - 10-20 mg per day or retabilic - 50 mg intramuscularly within 2-3 weeks.Apply iron preparations, but they can sometimes provoke hemolytic crisis. To prevent the cryosis, iron is prescribed in small doses against the background of treatment with anabolic steroids. During thrombosis, heparin is shown: with the first injection, 10,000 units are administered intravenously, then 5-10 thousand units 2-3 times a day under the skin of the abdomen (thin needle to a depth of 2 cm into fat tissue) under the control of blood coagulation. Contraindications for treatment of heparin are a recent aggravation of the ulcerative disease of the stomach or duodenum, as well as the presence of bleeding sources.
Paroxysmal night hemoglobinuria is a very rare disease from a group of hemolytic anemia, not considered inheritable. Acquired during life, although it has a genetic basis. The essence of pathology is changes in the structure of blood cells (most of all erythrocytes), leading to early destruction of their shell and intravascular decay (hemolysis).
The prevalence is about 16 cases by million of the population, and the annual incidence is 1.3 per million people from 20 to 40 years old, dependence on the floor was not detected.
The name includes the names of Italian researchers and doctors who have spent years to study: Markiafa-Mikeli disease, Strejbong-Markiafa.
What is hemoglobinuria, what is it caused?
Hemoglobinuria is a symptom of various diseases that cause the collapse of erythrocytes with its impact on the membrane, while hemoglobin leaves cells and enters the plasma.
A healthy person can have no more than 5% of the total blood plasma. Increased hemoglobin level of 20-25% is observed with congenital disorders or hemoglobinopathies (β-thalassemia, the destruction of red cells in sickle cell anemia).
Pronounced hemoglobinuria is caused by states when significantly exceed permissible norms Hemoglobin in connection with the hemolysis of red blood cells. The system of macrophages is not able to recycle such a large amount of pigment, and hemoglobin enters the urine.
The causes of hemoglobinuria can be:
- acute infectious disease (influenza);
- pneumonia;
- injuries;
- intoxication in poisoning with aniline dyes, carbolic acid, bertolen salt;
- sharp supercooling;
- strong and long-term physical stress;
- transfusion of different transparent blood;
- extensive burns;
- the role of the acquired mutation of the Pig-A gene is established.
Aniline dyes are widely used in the textile industry, the design of the batik, in the service of dry cleaning and dyeing, work with them requires caution
Hemoglobinuria does not happen without high level Hemoglobin in blood (hemoglobinemia). Predure paroxysms are associated with the physiological shift of acid-alkaline equilibrium towards the acidosis at night. The increased content of decay products even more contributes to the acidification of the body, enhancing the decay of blood cells.
Pathogenesis of violations
The main changes in paroxysmal night hemoglobinuria occur at the level of the complement. It is a chain of biochemical reactions that ensure innate immunity.
Active substance The educated membraneactive complex is considered. It contains about 30 regulators components. The synthesis of components of the complement depends on the signals obtained from the nervous and endocrine Systems. Normally, it is controlled by special proteins that do not allow to destroy the host cells (human).
With night hemoglobinuria, this process is lost. The lipid layer of the erythrocyte cell shell is destroyed, which causes their death. The increased sensitivity of the erythrocyte membrane to the complement component is proved.

Completement is necessary to protect cells from infectious agents and disposal of products of decay of microorganisms and their own damaged cells
Other blood cells (leukocytes and platelets) also react to the occurrence of defects in the shell. They did not find the accumulation of immunoglobulins, which proves the lack of an autoallergia mechanism and speaks in favor of the defeat of the overall predecessor cell. It is she who receives genetic information (order) about the destructive action.
The missing genetic portion of the stem cell is called GPI-AP. Its disadvantage in the erythrocyte clone contributes to the susceptibility to hemolysis under the influence of complement. At the same time, a normal clone of red blood cells can exist.
Paroxysmal night hemoglobinuria manifests itself only if the pathological clone prevails over normal. Erythrocytes from a clone with a partial or complete absence of GPI-AP are found in patients using flow cytometry. It is important that the number of pathological cells in patients is not the same.
Increased thrombosis during Markiafa-Mikeli disease is associated with the stimulation of blood coagulation liberated in the destruction of red blood cells factors.
Forms of the disease
Classification clinical shapes Consides laboratory data and causal relationships of blood changes. It is customary to allocate the following varieties:
- There is no subclinical - laboratory signs of hemolysis, only with highly sensitive methods you can reveal a small number of cells with the absence of a GPI-AP. There is no clinic of the disease. Often combined with aplastic anemia.
- Classical - all clinical symptomsThe periodic exacerbations occurs, in addition to the erythrocytes, leukocytes and platelets are affected, laboratory determines the signs of hemolysis (the growth of reticulocytes, serum enzyme lactate dehydrogenase, bilirubin, with a reduced Haptoglobin level). Blood-formal anomalies in the bone marrow is not observed.
- Caused by the insufficiency of bone marrowing at various diseases - It is assumed to be concomitant or suffered pathology of the bone marrow with impaired blood formation (with aplastic anemia, myelodysplastic syndrome). According to analyzes and clinics, all the manifestations of hemolysis are revealed against the background of bone marcing anomalies.
According to another classification, it is proposed to allocate:
- idiopathic shape or actually paroxysmal night hemoglobinuria;
- pathology in the form of syndrome under various diseases;
- rarely observed view occurring after bone marrow hypoplasia.
No classification is based on a quantitative indicator of the prevalence of an abnormal clone in the blood. It is shown that the subclinical flow is possible at 90% of the substitution of normal cells. And in other patients there are heavy thrombosis in the presence of only 10% of the altered erythrocytes.
Symptoms and clinical current
The disease can begin both suddenly (acutely) and have a gradual chronic course. Periods of exacerbations are called hemolytic crises. Often they are preceded by the transferred cold, communication with infection, contact with toxic substances.
The main symptoms of paroxysmal night hemoglobinuria include:
- stomach ache;
- pain in the chest of different intensity and localization - pain of different localization are associated with thrombosis of small branches of the arterial bed and the formation of foci of ischemia in the internal organs;
- signs of anemia (weakness, dizziness, headaches) - caused by increased destruction and insufficient products of erythrocytes, in addition, research indicates the deficit of iron and folic acid in the blood of patients;
- the jaggility of the skin and the scler is an indicator of the exit to the blood of a direct bilirubin, recycled with a hemoglobin surplus liver;
- swallowing;
- erectile dysfunction in men - manifests not only against the background of crises, but goes into chronic form, caused by a reduced nitrogen oxide concentration in plasma, impaired muscle and vascular tone.
- increased fatigue;
- shortness of breath, heartbeat;
- local signs of thrombophlebitis (redness of the skin area over veins, swelling, pain in palpation, temperature increase);
- when examining the patient, the doctor may noted the increased liver and spleen, this feature is especially important for diagnosing the development of thrombosis in them, heart attacks.
Chronic course of illness contributes to the development:
- light hypertension with thrombosis in the branches of pulmonary vessels;
- chronic renal failure caused by the deposition of the spree of hemoglobin (hemosiderin) in the renal tubules, vessel thrombosis with the formation of microindarkts;
- high sensitivity to joining infection.
These syndromes become the most likely reasons Fatal outcome.
Laboratory diagnostics
The diagnosis of Markiafa-Mikelet's disease is raised after a thorough examination in hematological centers with the possibility of conducting specific tests and tests.
In peripheral blood, detected:
- erytrogenia, leukopenia, thrombocytopenia (the state of oppression of the general sprout of blood cells is called pancytopenia);
- reticulocytosis;
- growth of hemoglobin in plasma;
- reduced iron and folate content.
When studying the bone marrow, they detect:
- signs of activation of erythropoese (production of erythrocytes) due to the accumulation of precursor cells (normosoblasts, plasma and fat cells);
- reduced amount of granulocytes and megacarocytes;
- sections of hemorrhages, accumulation of hemolyzed red blood cells in sinuses;
- at the stage of suppression of the blood formation, the zones of fatty rebirth are visible, emptying.
Specific tests based on the increased sensitivity of defective erythrocytes to the complement under the conditions most favorable in the composition of the medium are chem (acid) and Hartman tests (with sucrose).
Both samples check the "survival" of the erythrocytes of the blood sample, placed in a weak solution. The test of chem is positive when destroying 5% or more, and Hartman - 4% or more.
The Cumbas test is carried out to eliminate communication with the autoimmune mechanism for the destruction of cells, it is negative at night hemoglobinuria.

Urine coloring indicates a significant content in it oxymemoglobin
Study urine showed that one of the initial signs of night hemoglobinuria is the morning and night portion of urine, painted in a dark red color. Over time, the assembled urine is divided into layers:
- on top of a liquid transparent, but preserving staining;
- the particles of dead cells of organic origin are determined below.
What diseases should be distinguished by night hemoglobinuria?
Differential diagnosis of paroxysmal night hemoglobinuria is carried out with other similar clinical flow Anemias, primarily with the hemolytic aumethy of autoimmune type and aplastic.
General features are:
- sharp decrease in the number of erythrocytes;
- reticulocytosis;
- the presence of jaundice;
- fever;
- increasing the concentration of free bilirubin;
- tendency to thrombosis;
- moderate increase in liver and spleen.
With anemia there are no high numbers of hemoglobin in blood plasma, urobilin in the urine. Gem, Hartman laboratory tests are negative, but the Cumbas test is positive.
Diagnostics are significantly difficult if the disease occurs in the form of temporary crises against the background of an acute form of myeloblastic leukemia, erythromyelosis, osteomyelosclerosis, metastatic bone marrow damage for malignant tumors.

The erythrocyte mass is stored in the cold in special packages
Treatment
To date, there is no effective way to stop the decay of red blood cells. It remains only to use the replacement opportunity and overflow the patient washed with the erythrocyte mass of donors.
An important feature is a good "attitude" of the patient's body to administered alien cells, there is practically no reaction reaction. Given the presence in the shells of healthy GPI-AP cells and the absence of genetic mutations in them, it is possible to maintain the patient's blood formation.
Blood used for transfusion should be stored in a frozen state of at least a week in order to complete leukocyte in it. Having come to the patient, they may cause hemolysis exacerbation due to increased sensitization and complement activation.
With frequent transfusions, the formation of antioeritrocyte antibodies is possible. Such a patient, subsequent transfusion is carried out after several procedures for laundering erythrocytes with saline and testing donor blood using the Cumbas reaction.
The number of transfusions is usually assigned at least five, but depends on the severity of the patient's condition and reaction to treatment.
To stimulate the correct hemopower use neopol (anabolic hormonal drug) courses up to three months. It is possible to change functional state Liver.
In order to treat and prevent thrombosis, heparin is used, followed by the transition to the supporting doses of indirect anticoagulants.
To compensate for iron losses, preparations are prescribed in tablets.
Indication to the removal of the spleen can be a sharp increase, signs of heart attack. Splenectomy is rare.
Preparations of hepatoprotective action are prescribed to protect the liver. Sometimes steroid therapy helps.

The drug is administered only intravenously drip
In recent years, information about the application has appeared. medicinal preparation Echulizumab (soliris) made of monoclonal antibodies. Judging by the reports available, it blocks hemolysis, is able to confront the complement of the blood. The drug is considered the most expensive medicine in the world. His action and negative effects are not sufficiently studied.
Night hemoglobinuria has not yet have specific treatment. Even with sufficient supporting therapy, patients live about five years after the start of the disease. Prevention does not exist. Everyone should adhere to proper behavior when working and forced contact with toxic compounds.
Night hemoglobinuriaParoxysmal night hemoglobinuria (PNG) (Markiafa-Mikelet's disease) - a rarely occurring acquired disease, characterized by chronic hemolytic anemia, mixed or permanent hemoglobinuria and hemosideurine, thrombosis phenomena and cytopenia of peripheral blood and bone marrow aplasia.
APG - a clonal bone marrow disease arising from mutation of a blood stem cell.
The consequence of the mutation is the appearance of a clone of anomalous hemopoietic cells with a defect of a cytoplasmic membrane, making them improvedly sensitive to the complement in an acidic environment.
Epidemiology.
For 1 year, 2 new APG cases are revealed for 1 million people.
Faces are somewhat more often ill, regardless of gender.
Etiology PNG studied not enough.
The proven clonalness of the disease suggests the role of etiological factors that can cause a mutation of a blood stem cell.
Pathogenesis.
Due to the mutation of the blood stem cell in the bone marrow, a pathological clone of cells is developing, which gives rise to abnormal erythrocytes, granulocytes and platelets.
At the genetic level, the cause of the Development of the PNG is the point mutation of the PIG-A gene (from English. PhosphatiDilinositol Glycan Complementation Class A) associated with an active X chromosome.
This mutation leads to a violation of the synthesis of glycosylphosphatidyl alternate anchor (HPFE anchor), which is responsible for attaching a series of membrane proteins to the cell membrane.
The descendants of the mutated stem cell acquire a special sensitivity to complement due to the restructuring of the structure of their cytoplasmic membrane.
As a clonal disease, APG can be transformed into Onell (most often in acute erythroblastic leukemia) and MDS.
Clinical picture.
In most cases, the disease is manifested by symptoms of anemia: weakness, increased fatigue, shortness of breath, heartbeat, dizziness.
Hemolytic crises can be provoked by infection, severe physical activity, surgical interventions, menstruation, drug intake.
In the classical form of the disease, the hemolytic crisis is usually developing at night when there is some blood pH in the acidic side.
In this case, after the awakening there is urine of black. Hemoglobinuria is usually considered as a characteristic feature, however, it is not mandatory.
A more permanent symptom - hemosideuria.
Hemolysis can be accompanied by pains in lumbar, dye areas, bones and muscles, nausea and vomiting, fever.
In case of inspection: Pallor, jaggility, bronze skin and splenomegaly.
Gemolytic crises are replaced by a relatively calm condition, in which the degree of hemolysis and the severity of clinical manifestations decrease.
Patients are prone to thrombosis, leading to death (thrombosis of brain vessels, coronary vessels of the heart, large vessels abdominal cavity and liver).
The occlusion of the glomerular kidney is the cause of the OPN.
Excessive accumulation of iron in the kidneys (hemosiderosis) can cause CPN.
Diagnostics.
The primary representation of the APG is to develop when identifying clinical and laboratory signs of intravascular hemolysis: Normochromic anemia; an increase in the number of reticulocytes; The appearance of black urine (hemoglobinuria); Hemosiderin detection in the urine; Pain in the lumbar and left-hand area.
APG suspected increases with a combination of all the above-mentioned signs with changes in the hemogram: normal anemia, leukopenia and thrombocytopenia are detected.
As the disease develops, anemia is acquiring a hypochromic nature due to significant losses of iron with urine.
The identification of repeated thrombosis also helps in diagnosis. The absence of signs of intravascular hemolysis (for example, in the intercrown period) makes it difficult for the APG diagnosis.
In such a situation, it is possible to suspect the existence of the disease only with careful analysis of anamnestic data.
To confirm or eliminate the PNG, you must perform a trial of chem and (or) sucrose sample.
These tests make it possible to identify the increased sensitivity of the erythrocytes to the complement in the acidic medium, even during the absence of clinical manifestations of intravascular hemolysis.
A positive result obtained when performing any of the specified tests is a necessary and sufficient condition for statement of APG.
The negative results of both tests exclude APG diagnosis.
Relatively recently, a flow cytofluorimetry method based on the determination of reduced expression of CD-55 and CD-59 on erythrocytes and leukocytes, CD-14 on monocytes, CD-16 on granulocytes and CD-58 on the lymphocytes began to be used to verify the diagnosis of APG.
According to some data, this method for its reliability is not inferior to the above samples.
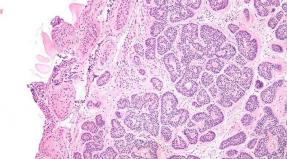
Bone marrow pathorphology: moderate hyperplasia Erythitoid sprout of blood formation.
The number of granulocyte predecessors and megalocytes is reduced.
In some cases, fields of pronounced hypoplasia, represented by edema stroma and fat cells, can be detected.
Differential diagnosis.
When identifying signs of intravascular hemolysis, the diagnosis is not relieved, since the circle of diseases with intravascular hemolysis is limited.
Difficulties in this group can meet only when delimiting the PNG and AIG with thermal hemolyns.
Both diseases are very similar in the clinical picture, but the number of leukocytes and (or) platelets are often reduced at APG.
Doubts are completely allowed by the immunological methods of detection of AT class hemolysins in the serum of patient and (or) fixed on the surface of anti-rasty anti-crocyte AT.
In all cases, the detection of intravascular hemolysis seems to be appropriate to include the sucrose sample and (or) sample of the chem in the number of priority tests.
When the cytopenia and the absence of clinical and laboratory signs of intravascular hemolysis, there is a need for differential diagnosis with diseases accompanied by pancetopenia in peripheral blood, namely aplastic anemia and myelodic syndrome.
Performance cytological research Bone marrow will make it slightly narrowing the range of differential diagnostic search.
Treatment.
The pathogenetic method of treating APG is bone marrow transplantation from a histo-compatible donor.
If it is impossible to choose a donor for myelotransplantation, then symptomatic therapy is carried out, aimed at the relief of emerging complications, and replacement therapy with washes of erythrocytes or frostably washed red blood cells.
The use of prednisolone or other GCS and (or) immunosuppressants does not give effect due to the lack of an application point for the data of these drugs.
Forecast.
The life expectancy of patients averages 4 years.
Cases of long spontaneous remission are described.
Prevention.
Effective PNG prevention does not exist.
Heparin is assigned to the prevention and treatment of thrombosis.
Reopolyglyukin also apply to improve rheology.
In this group of patients, there is no family trend towards anemia, there are no concomitant innate anomalies and there are no violations in the neonatal period. Aplastic anemia may occur at any age in children and in adults, it can sometimes be associated with specific intoxication or infection, but often such a connection is not observed and the anemia is considered "idiopathic".
Some medications, such as 6-mercaptopurine, methotrexate, cyclophospham and busulfan, have a certain, pre-predictable dose-dependent ability to coal bone marrow. If this oppression will continue, it will lead to an aplasia of the bone marrow, which usually goes quickly after the discontinuation of the drug. These medicines damage normal bone margins by means of the same mechanism that when they suppress the growth of leukemic cells. The biochemical principles of their action are quite well studied. By the same category relates radiation damage to the bone marrow.
Other medicines, such as Akrichin, Chloramphenicol, Phenylbutazone and anticonvulsant drugs used in normal healing doses, can cause deep aplasia of the bone marrow in a very small number of people, and it is impossible to predict this aplasia. Often it is irreversible and about half of the patients die. The same category includes intoxicating insecticides, such as DDT, and some organic solvents. It often remains unclear whether to connect anemia with one or another medicine. A prerequisite for such a connection is to receive medicines during the last 6 months. The most famous and studied from them chloramphenicol. This drug stands at the head of the list of known etiological agents in a group of patients with acquired aplastic anemia described by Scott and co-authors, and in the same groups of sick children from Shahidi. Gourmet watched in Sydney for 8 years 16 cases in which the disease was connected, as expected with the reception of chloramphenicol. The absolute frequency of deathly acquired aplastic anemia in populations, which did not know about contacts with any dangerous medicine and was known to contact with various medicines, including chloramphenicol.
Treatment of chloramphenicol increases the likelihood of the development of aplastic anemia 13 times, but it is also clear that this growth is small. For other medicines, the risk is even less. Nevertheless, the British Committee for the Safety of Medicines recommends with all diseases, in addition to abdominal typhoids and hemophilic influenza meningitis, apply chloramphenicol systemically, only after a thorough clinical and usually laboratory study indicating that another antibiotic will not be enough. Never use it systemically at a banal infection.
The mechanism for the development of aplastic anemia under the action of chloramphenicola is unclear. The emergence of aplastic anemia is not associated with the dose and duration of treatment, it can also be explained by insufficient excretion from susceptible to individuals. In vitro One can prove the suppression of nucleic acid synthesis in normal bone marginal cells, but only with such a concentration of the medication, which exceeds the in vivo used. There is an assumption that small amounts of chloramphenicol can be consumed with milk from cows treated for mastitis, that these small quantities can sensitize the bone marrow to therapeutic doses applicable in the future. It was also assumed that there is no unopened synergism with other medicines, probably harmless if they are applied separately. Discussing the etiology of a pancytopenic lethal aplasia caused by chloramphenicol, it should be noted that a significant part of patients receiving this medication observed a completely different, reversible and dose-dependent oppression of the bone marrow. In 10 of the 22 patients receiving chloramphenicol, multiple large vacuoles were found in the early erythroblasts of the bone marrow, which was often accompanied by a drop in the number of erythrocytes and reticulocytes. These changes disappear a week after the cancellation of the medication. The development of them, apparently, contributes to elevated doses, slowed down clearance from plasma and accelerated erythropoes. The same vacuoles can be seen with phenylalanine or riboflavin deficiency.
With regard to the etiology of other drug aplasia, there was always a temptation to assume the effect of immune mechanisms, perhaps the type of medication - Hapten. However, these mechanisms have never been demonstrated. Only in one clinical situation, namely, during the reaction, the transplant against the owner in immunologically insolvent infants who received transfusion has established an immunological origin of aplastic anemia. The development of a sharply expressed anaphylactoid reaction after accidental re-contact with DDT in a sensitive patient also suggests the immune mechanism. Newvig offered three explanations of drug aplasia: a) direct and toxic action on bone marginal cells, for example, after chronic production contact with benzene; b) True allergies, the manifestations of which arise quickly after contact with a low dose; c) Long contact with large doses, i.e. "Allergy of high doses." This is the most common form. The author explains it primarily by damage to cell membranes. It can also assume a genetic predisposition, which indicates the case of blood discrasion after contact with chloramphenicol in single-time twins. Recently, review articles on drug aplastic anemia in the magazine "LanCet" were recently published.
Similar problems arise in connection with the preceding development of aplastic anemia with viral infection. This phenomenon is well studied with infectious hepatitis. Aplastic anemia in 5 patients aged 4 to 19 developed 1-7 weeks after the start of hepatitis. A number of similar cases are described, including 3 cases of Schwartz and co-authors. These authors noted that with infectious hepatitis, a temporary decrease in the number of granulocytes, platelets and hemoglobin is often observed and that progressive changes leading to bone marrow aplasia, in a very small number of patients may be a continuation of the entire process that is probably probably from a genetic predisposition. Here you can see an analogy with intoxication by chloramphenicol. Pancitopenia with temporal hypoplasia of the bone marrow is also described in connection with a number of infections caused by RNA-containing viruses, including rubella viruses and influenza microviras, paragripping viruses, pig viruses and measles. Two experimental viral infections in mice, i.e. MVH-3 and the Trinidadian strain of Venezuelan horsepower encephalitis, cause bone marrowing and bone marrow hypoplasics, and a virus can be seized from bone marrow. As in other reasons of aplastic anemia, an autoimmune process is assumed.
At about half of the cases of acquired aplastic anemia, it is impossible to detect a history of a serious preceding infection or contact with toxic agents. Wolf published a large material that includes 334 cases of the acquired shelling, and in 191 cases, i.e., 57.2%, anemia is recognized as idiopathic.
In the material of the gourmet, the relative number of patients with idiopathic anemia was less, that is, 28 out of 104, who suffered acquired by Aplasia. In 5 out of 17 cases, according to the materials of Shahidi and in 5 out of 9 cases, the materials of the referee anemia was idiopathic. It is not yet clear whether diseases are caused in these cases by an infections of an unidentified virus. At least some of the idiopathic cases seems to be allocated in special groupwhich could be called preykelosis or leukemia in the aplastic phase.
Melkhorn and co-authors describe 6 children who, on the basis of strong, indisputable evidence, was diagnosed with aplastic anemia aged 1 year 11 months to 6 years, but all these children in the future, after 9 weeks - 20 months, developed acute lymphoblastic leukemia. . These 6 patients had one common feature - faster than usual, the therapeutic effect in comparison with aplastic anemia for initial therapy with corticosteroids. The same noted Gourmet, and we also observed this effect in one case, in which acute lymphoblastic leukemia developed in 3 months. This rapid reaction to treatment only by one corticosteroids is noticeably different from the usual lack of effect in other cases of aplastic anemia. It should be noted that a similar leukemic transformation of aplastic anemia caused by benzene and chloramphenicol is described.
Symptoms of purchased aplastic anemia
Acquired aplastic anemia is characterized by approximately the same symptoms and objective signs, as well as a constitutional form, but there are no pigmentation, low growth and congenital skeleton anomalies or internal organs. The age framework, in which the disease occurs, is wider, except, be an aplasia caused by chloramphenicol, in which the "peak" of the maximum morbidity lies between the 3rd and 7th year. In 43;% of patients with an acquired form of illness in the large material Wolf: and 67% in the large summary material gourmet in history was a contact, sometimes repeated, usually over the preceding 6 months, with medicines or chemicals, relative to which they are known that they They predispose to aplastic anemia.Newman and co-authors described 14 children with idiopathic pancetopenia and noted that, apart from the three main signs - anemia, fever and purples, there are important negative features, that is, the absence of hepatoslenomegali, lymphoacident, ulcers in the oral cavity and jaundice. However, it is possible to observe the purple of the mucous membrane and bleeding from the gums. Sometimes there may be inflammatory lymphadenopathy associated with local sepsis.
If the child appears red urine, then the development of paroxysmal night hemoglobinuria should be assumed.
Laboratory diagnostics
The picture of peripheral blood is about the same as with a constitutional form, but neutropenia is deeper, sometimes approaches agranulocytosis. In addition, there is a more pronounced aplasia of the bone marrow, which consists of almost some of the fatty sections devoid of hemic cells. In 5-90% of the erythroid predecessors, which still have in the bone marrow, there are megaloblastic changes and other signs of "dierythropois". In patients with inverse bubbling of bone marrow associated with the dosage and caused by chloramphenicol, in the bone marrow, there is a vacuolaization of erythroid and myeloid predecessors, similar to the fact that it is tinned to observe with phenylalanine deficiency. The level of fetal hemoglobin can be increased to the same measure as with constitutional forms, but less constant. It was assumed that the levels above 400 μg% (or 5%) indicate a more favorable forecast in the case of acquired disease, but the analysis of later cases treated at the same institute did not confirm this data, possibly due to the use of another method.Aminaciduria, observed about half of patients with a constitutional form, there is no and no lag behind bone age.
More than half of adult patients suffering from this disease, lymphopianic and hypogammaglobulinemia with a subnormal level of IgG were found.
Related hemolysis, including paroxysmal night hemoglobinuria. Part of the patients with aplastic anemia is shortened by the life expectancy of red blood cells. This suggests that the erythrocyte defect sometimes happens not only quantitative, but also qualitative. In this case, you can observe increased sequestration in the spleen. Reticulocytosis, which would have to be, usually excluded due to the aplasia of the bone marrow. In some cases, the content of Gaptoglobin is reduced. One of the causes of hemolysis in this disease is an unusual syndrome of a combination of paroxysmal night hemoglobinuria (PNG) and aplastic anemia. This syndrome must be assumed when the patient with aplastic anemia increases bilirubin or spontaneous reticulocytosis appears. The diagnosis is confirmed by the test of acid serum hemolysis (KSG) on the APG, as well as tests for hemosideurine. In some cases, the APG can only be found in the study of the most sensitive erythrocyte population, i.e., reticulocytes and young erythrocytes obtained with a thorough removal of a layer pipette below the leukocyte-thrombocyte clock after centrifugation of 20-35 ml of blood at 500 g.
Usually, at the same time, the APG syndrome is detected against the background of aplastic anemia, often after the ErythropoEz recovered to a certain extent. In several cases there was a reverse sequence, that is, on the background of the PNG, severe or fatal insufficiency of bone marrow developed. Levis and Dace systematically set tests in all of their patients with aplastic anemia and found that 7 out of 46 (15%) have a Lab Criteria for APG. In 2 of them, a typical PHP picture was developing. Applying to this issue from another point of view, the authors found that at least 15 of the 60 patients with APG initially had signs of aplasia. Usually APG is a disease of adult men. However, the form that occurs during aplasia, apparently, is found in a younger age and can affect children. Gardner watched 11 such patients, including 6 to 25 years, 2 patients were 7 and 9 years old. These two were boys. They have an aplastic anemia before the diagnosis of PNG lasted 2 years and 5 years.
An interesting feature of this combined syndrome is that aplastic anemia can be type Fanconi can be acquired after contact with chloramphenicol, tranquilizers, insecticides, herbicides and other substances, and maybe idiopathic. Levis and Dace believe that the main bond exists between the aplasia of the bone marrow and APG, and not between the etiological factors caused by bone marrow damage and APG. Both of these author, as well as Gardner and Bloom, suggest that in the period of Aplasia there is a somatic mutation of bone marginal stem cells, which leads to the emergence of a secondary clone of pathological erythrocytes inherent in APG, which begin to produce during the subsequent regeneration of the bone marrow. It should be added that, although the characteristic defect with APG is concentrated in red blood cells, granulocytes are also changed. The "skin window" method shows a decrease in their phagocytic activity and alkaline phosphatase activity. In contrast, with an uncomplicated aplastic anemia, alkaline phosphatase activity in granulocytes is usually increased.
Treatment
Treatment in principle is the same as with a constitutional aplastic anemia, however, it is necessary to make sure to stop all contacts with a medicine or toxic agent if it is known. Repeated contact can cause lethal relapse in patients who survived the first attack of aplasia, and may even provoke a lethal anaphylactic shock.Supporting measures also include blood transfusions, while anemia is deep enough to cause symptoms, usually at the level of hemoglobin 4-6 g%. The erythrocytic mass is applied not only for the treatment of obvious bleeding, and it is necessary to strive to increase the level up to 8-9 g%. A higher level of hemoglobin leads to a sharper depression of erythropoese. Thrombocytopenic bleeding is treated with rapidly enriched plasma-enriched plasma or platelet concentrates (4 units / m2). Intramuscular injections should be avoided. With all the procedures, it is necessary to strictly observe the aseptics and vigorously treat infections with bactericidal antibiotics. Since neutropenia is usually particularly deep with the forms of aplastic anemia, during the neutropenic phase, it is possible to use a special neutropenic regime: rinsing the oral cavity is 0.1% hybitanium solution 4 times a day after meals (made of pure antiseptic without detergents and dyes); lubrication by the inseptic ointment nostrils 3 times a day; Daily bath. Lubrication gums 1% Hibitan dental gel 2 times a day (instead of brushing teeth cleaning). When patients are in the hospital, some kind of isolation with a reversing barrier is needed to reduce the danger of infection with a microflora. Preventive systemic antibiotic therapy should be absolutely rejected, as it increases the tendency to fungal infections and infections resistant to antibiotics. Starting infection can manifest itself an increase in the tendency to bleeding. In case of infection, the number of platelets is not only reduced, but also increases a hemorrhagic trend at a given number of platelets.
Androgen. The specific therapy androgens + corticosteroids are carried out in the same way as with constitutional forms, i.e. oxymetalone inward - 4-5 mg / kg per day + prednisone 5 mg 2 times a day in children with a body weight up to 20 kg, 5 mg 3 3 times per day with a body weight from 20 to 40 kg and 4 times a day with a body weight of more than 40 kg. The difference lies in the fact that when the forms of anemia acquired, the effect is achieved in a smaller percentage of patients, the reaction to treatment is slower, but remission in patients who can be treated continues after canceling androgen and corticosteroids. With anemia of FanConi, bone marrow deficiency quickly recurns after the termination of this therapy. It was even indicated that this circumstance could be used in difficult cases in differentiation of the acquired form from constitutional.
The first results of treatment with androgen and steroids were very impressive. Of 17 children with acquired aplastic anemia (in 12 cases, the toxic, in 5 - idiopathic) in 10 was observed with a resistant reticulocytosis, which reached a vertex of 5-15% after 1-7-month combined treatment with androgen and corticosteroids. Of these children, 9 survived, and in the future, the content of hemoglobin has increased. In 3 children there was transient reticulocytosis without other reactions. The discrepancy between the deadlines for the appearance of reticulocytosis and the increase in hemoglobin in these patients was explained by hemolysis. In addition, red blood cells, which are formed in the early stage of the regeneration of the bone marrow, hypochroms at a normal level of iron in serum and elevated, the content of free protoporphyrin in the erythrocytes, which indicates the cellular cell of the hemoglobin synthesis. The maximum increase in hemoglobin was observed 2-15 months after the start of treatment with androgen. In the study of the bone marrow in the dynamics in the early stage of treatment, groups of reticular cells were found, which ripen and turn into erythroid foci, in those patients who later develop a reaction to treatment. In all patients with increasing hemoglobin, an increase in the number of segmented cells was also observed in more than 1500 in 1 μl, but the platelet reaction was less pronounced and they achieved only 25,000-90,000 in 1 μl. Typically, the number of segmented neutrophils increased more slowly than the level of hemoglobin, the number of platelets increased even slower. The total duration of treatment with androgens in these patients ranged from 2 to 15 months, after the cessation of treatment, they remained the remission of vaguely long. In 2 positively responding to the treatment of patients there was an idiopathic aplasia, in 8 - toxic. Among the patients who did not react, 3 had an idiopathic and 4-toxic form of aplasia. The authors suggested that long-term treatment of high doses of corticosteroids could disrupt the bone marrow function due to an increase in the number of adipose tissue in the bone marrow.
Similar results were assessed and co-authors who used androgens + steroids. In 5 out of 9 children with an acquired aplastic anemia, a pronounced hematological improvement has come, which turned out to be stable. 2 children had idiopathic and 3 - toxic form. (From patients who did not respond, 3 had idiopathic and 1-toxic anemia.) A similar relationship ratios were observed. The number of platelets increased significantly only 9-17 months after the start of treatment, and even then it reached only 50,000 in one patient and 100,000 in 1 μl in 2 patients, while hemoglobin and segmented cells were normal. Treatment stopped after 7-11 months; In 4 out of 5 patients, the hemoglobin level was temporarily fell by 1-3 months. Patients were supervised from 1 year to 3 years. During this time they had no relapses.
According to these two messages, a positive reaction was observed by a little more than half of children, and the treatment was effectively both in idiopathic and toxic forms of aplastic anemia. Among patients with toxic forms, the reaction frequency was, maybe somewhat larger.
Until the last of these articles appeared, it was the impression that without treatment with androgens, patients rarely survive. The increased survival noted in the two most recent reports is explained by the success of symptomatic therapy, including platelet antibiotics and transfusion. In particular, Hane and co-authored material sheds a new light on the natural course of the disease and, apparently, fills the gap between patients treated androgens, and without androgens (in 30 of 33 patients, the ethiology of anemia was toxic, and not idiopathic, which may be Explains a more favorable forecast). Gourmet in a review covering 104 patients with acquired aplastic anemia from Boston and Sydney indicates that the overall survival was equal to 34% in the combined treatment with androgens and corticosteroids and 19% when treating only corticosteroids or supporting therapy.
In newer messages, including the results obtained in the same Boston Children's Hospital, the data is less satisfactory. Mortality was 70-80%, despite androgens, corticosteroids and supporting therapy. Two-phase survival curve. Many patients in the early period die from infections and bleeding during the first 6 months. Currently, the effectiveness of androgens in patients with severe acquired aplasia is questioned.
Prognostic signs. According to the work of the gourmet, the forecast seems to be worse in aplastic anemia after infections, especially infectious hepatitis, or after one short course of chloramphenicol. The forecast is better in idiopathic cases, as well as in patients with anemia, which can be explained by the reception of anticonvulsant or repeated courses of chloramphenicol. It was assumed that the bone marrow of the child who had an aplastic anemia after one short course, often more oppressed than a child who had barcitops only caused by repeated courses of medicines. It is known that children with a sharp hypocloma of bone marrow on a particularly difficult forecast indicates the number of lymphocytes of more than 85% in the bone marrow, the number of neutrophils is less than 200 in 1 μl or platelets less than 20,000 in 1 μl. Based on these data, Hamitt and co-authors suggested that severe aplasia after hepatitis should be considered as an indication for the early bone marrow transplant due to the fact that only about 10% of patients of this kind survive with supporting therapy + + androgens and steroids.
Bone marrow transplantation. Due to the failure of treatment with androgens of severe acquired aplastic anemia, researchers turned to the possibilities of the prospects for bone marrow transplantation. After intravenous bone marrow injections from single-line twins in 5 out of 10 cases fast recovery Bone marrow functions. If there are no donors in the form of single-time twins, then a serious obstacle is the possible rejection of the transplant or, if it takes place, the transplant reaction against the host. However, among conventional Sibs there is one chance of 4, which will be found a histociable donor, selected by typing HL-A and mixed lymphocytic culture to identify other histocompatibility lockers. These precautions reduce the value of the problem of incompatibility of the transplant, but do not solve it completely. In order to reduce or eliminate the possibility of rejection, additional immunosuppressive therapy requires, for example, high doses of cyclophosphamide before the bone marrow transplant and the methotrexate course after a transplant. Before trying to carry out this therapeutic measure, it is necessary to carry out massive supporting therapy, including the patient's detection in a sterile medium, leukocyte and platelet transfusion during the critical first days, as well as the presence of a medical team with extensive experience. Thomas and co-authors describe the technique of fence, processing and infusion of bone marrow. 24 patients (including 8 under-14 years old) with severe aplastic anemia (14 cases of idiopathic anemia, 4 cases of anemia after hepatitis, 4 - drugs, 1 - PHG, 1 - anemia of Fanconi), which were not amenable to conventional treatment, received grafts from Sibs, identical by HL-A. In 21 patients, the bone marrow regeneration was observed, which in most cases, as was established using genetic markers, was due to donor cells. In 4 patients, the transplant was rejected and they died. Four patients died from secondary illness, 11 people live with functioning grafts. Observation period from 141 days to 823 days. Ten patients returned to a normal active lifestyle. These results obtained by the Siethla researchers group prompted others to use this method. In fig. 25 shows the result of a transplant in the first case in the UK, carried out by a bone marrow transplant brigade at the Royal Marsden Royal Hospital. It is possible that this will be the further treatment of individual patients with bad prognostic signs at the first appeal for help.
Various types of treatment. Patients who cannot be other treatment with a cell bone marrow shown splectomy. However, the intended effect of this operation was not confirmed when analyzing a large group of cases, and, since splenectomy is quite dangerous in these patients with thrombocytopenia, it is generally not recommended. Patients with hemolysis element and with sequestration of red blood cells in the spleen were possible. It has been established that splenectomy increases the lifespan of platelets in patients with aplasia, which ceased to help the transfusion of platelets.
In aplastic anemia, it was proposed to introduce intravenously phytohemagglutininin, but currently assembled data do not support the assumptions about the feasibility of this method. Iron treatment is contraindicated, as well as the treatment with cobalt causing nausea, vomiting and increase thyroid gland. Folic acid and vitamin B12 are ineffective even in patients with megaloblastic changes.
WWW Women's Journal.
The essence of pathology is changes in the structure of blood cells (most of all erythrocytes), leading to early destruction of their shell and intravascular decay (hemolysis).
The prevalence is about 16 cases by million of the population, and the annual incidence is 1.3 per million people from 20 to 40 years old, dependence on the floor was not detected.
The name includes the names of Italian researchers and doctors who have spent years to study: Markiafa-Mikeli disease, Strejbong-Markiafa.
What is hemoglobinuria, what is it caused?
Hemoglobinuria is a symptom of various diseases that cause the collapse of erythrocytes with its impact on the membrane, while hemoglobin leaves cells and enters the plasma.
A healthy person can have no more than 5% of the total blood plasma. Increased hemoglobin level of 20-25% is observed with congenital disorders or hemoglobinopathies (β-thalassemia, the destruction of red cells in sickle cell anemia).
The causes of hemoglobinuria can be:
- acute infectious disease (influenza);
- pneumonia;
- injuries;
- intoxication in poisoning with aniline dyes, carbolic acid, bertolen salt;
- sharp supercooling;
- strong and long-term physical stress;
- blood disease;
- transfusion of different transparent blood;
- extensive burns;
- the role of the acquired mutation of the Pig-A gene is established.
Aniline dyes are widely used in the textile industry, the design of the batik, in the service of dry cleaning and dyeing, work with them requires caution
Hemoglobinuria does not happen without a high level of hemoglobin in the blood (hemoglobinemia). Predure paroxysms are associated with the physiological shift of acid-alkaline equilibrium towards the acidosis at night. The increased content of decay products even more contributes to the acidification of the body, enhancing the decay of blood cells.
Pathogenesis of violations
The main changes in paroxysmal night hemoglobinuria occur at the level of the complement. It is a chain of biochemical reactions that ensure innate immunity.
The active substance is considered an educated membranage complex. It contains about 30 regulators components. The synthesis of components of the complement depends on the signals obtained from the nervous and endocrine systems. Normally, it is controlled by special proteins that do not allow to destroy the host cells (human).
With night hemoglobinuria, this process is lost. The lipid layer of the erythrocyte cell shell is destroyed, which causes their death. The increased sensitivity of the erythrocyte membrane to the complement component is proved.
Complete is necessary to protect cells from infectious agents and disposal of products of decay of microorganisms and its own damaged cells
Other blood cells (leukocytes and platelets) also react to the occurrence of defects in the shell. They did not find the accumulation of immunoglobulins, which proves the lack of an autoallergia mechanism and speaks in favor of the defeat of the overall predecessor cell. It is she who receives genetic information (order) about the destructive action.
The missing genetic portion of the stem cell is called GPI-AP. Its disadvantage in the erythrocyte clone contributes to the susceptibility to hemolysis under the influence of complement. At the same time, a normal clone of red blood cells can exist.
Paroxysmal night hemoglobinuria manifests itself only if the pathological clone prevails over normal. Erythrocytes from a clone with a partial or complete absence of GPI-AP are found in patients using flow cytometry. It is important that the number of pathological cells in patients is not the same.
Increased thrombosis during Markiafa-Mikeli disease is associated with the stimulation of blood coagulation liberated in the destruction of red blood cells factors.
Forms of the disease
The classification of clinical forms takes into account the laboratory data and the causal relationship of blood changes. It is customary to allocate the following varieties:
- There is no subclinical - laboratory signs of hemolysis, only with highly sensitive methods you can reveal a small number of cells with the absence of a GPI-AP. There is no clinic of the disease. Often combined with aplastic anemia.
- Classical - there are all clinical symptoms, flows with periodic exacerbations, besides erythrocytes, leukocytes and platelets are affected, laboratory decisions of hemolysis (growth of reticulocytes, whey enzyme lactate dehydrogenase, bilirubin, with a reduced Haptoglobin level). Blood-formal anomalies in the bone marrow is not observed.
- Caused by the insufficiency of bone marrowing at various diseases - is assumed to be concomitant or transferred by bone marrow pathology with impaired blood formation (in aplastic anemia, myelodsplastic syndrome). According to analyzes and clinics, all the manifestations of hemolysis are revealed against the background of bone marcing anomalies.
According to another classification, it is proposed to allocate:
- idiopathic shape or actually paroxysmal night hemoglobinuria;
- pathology in the form of syndrome under various diseases;
- rarely observed view occurring after bone marrow hypoplasia.
Symptoms and clinical current
The disease can begin both suddenly (acutely) and have a gradual chronic course. Periods of exacerbations are called hemolytic crises. Often, the transferred cold is preceded by them, contact with infection, contact with toxic substances.
The main symptoms of paroxysmal night hemoglobinuria include:
- stomach ache;
- pain in the chest of different intensity and localization - pain of different localization are associated with thrombosis of small branches of the arterial bed and the formation of foci of ischemia in the internal organs;
- signs of anemia (weakness, dizziness, headaches) - caused by increased destruction and insufficient products of erythrocytes, in addition, studies indicate the deficiency of iron and folic acid in the blood of patients;
- the jaggility of the skin and the scler is an indicator of the exit to the blood of a direct bilirubin, recycled with a hemoglobin surplus liver;
- swallowing;
- erectile dysfunction in men - manifests not only against the background of crises, but passes into a chronic form, caused by a reduced nitrogen oxide concentration in plasma, impaired muscle and vascular tone.
- increased fatigue;
- shortness of breath, heartbeat;
- local signs of thrombophlebitis (redness of the skin area over veins, swelling, pain in palpation, temperature increase);
- when examining the patient, the doctor may noted the increased liver and spleen, this feature is especially important for diagnosing the development of thrombosis in them, heart attacks.
Chronic course of illness contributes to the development:
- light hypertension with thrombosis in the branches of pulmonary vessels;
- chronic renal failure caused by the deposition of the spree of hemoglobin (hemosiderin) in the renal tubules, vessel thrombosis with the formation of microindarkts;
- high sensitivity to joining infection.
These syndromes become the most likely causes of fatal outcome.
Laboratory diagnostics
The diagnosis of Markiafa-Mikelet's disease is raised after a thorough examination in hematological centers with the possibility of conducting specific tests and tests.
In peripheral blood, detected:
- erytrogenia, leukopenia, thrombocytopenia (the state of oppression of the general sprout of blood cells is called pancytopenia);
- reticulocytosis;
- growth of hemoglobin in plasma;
- reduced iron and folate content.
When studying the bone marrow, they detect:
- signs of activation of erythropoese (production of erythrocytes) due to the accumulation of precursor cells (normosoblasts, plasma and fat cells);
- reduced amount of granulocytes and megacarocytes;
- sections of hemorrhages, accumulation of hemolyzed red blood cells in sinuses;
- at the stage of suppression of the blood formation, the zones of fatty rebirth are visible, emptying.
Both samples check the "survival" of the erythrocytes of the blood sample, placed in a weak solution. The test of chem is positive when destroying 5% or more, and Hartman - 4% or more.
The Cumbas test is carried out to eliminate communication with the autoimmune mechanism for the destruction of cells, it is negative at night hemoglobinuria.
Urine coloring indicates a significant content in it oxymemoglobin
Study urine showed that one of the initial signs of night hemoglobinuria is the morning and night portion of urine, painted in a dark red color. Over time, the assembled urine is divided into layers:
- on top of a liquid transparent, but preserving staining;
- the particles of dead cells of organic origin are determined below.
What diseases should be distinguished by night hemoglobinuria?
Differential diagnosis of paroxysmal night hemoglobinuria is carried out with other, similar to the clinical course anemia, primarily with the hemolytic anemia of autoimmune type and aplastic.
General features are:
- sharp decrease in the number of erythrocytes;
- reticulocytosis;
- the presence of jaundice;
- fever;
- increasing the concentration of free bilirubin;
- tendency to thrombosis;
- moderate increase in liver and spleen.
With anemia there are no high numbers of hemoglobin in blood plasma, urobilin in the urine. Gem, Hartman laboratory tests are negative, but the Cumbas test is positive.
Diagnostics are significantly difficult if the disease occurs in the form of temporary crises against the background of an acute form of myeloblastic leukemia, erythromyelosis, osteomyelosclerosis, metastatic bone marrow damage for malignant tumors.
The erythrocyte mass is stored in the cold in special packages
Treatment
To date, there is no effective way to stop the decay of red blood cells. It remains only to use the replacement opportunity and overflow the patient washed with the erythrocyte mass of donors.
Blood used for transfusion should be stored in a frozen state of at least a week in order to complete leukocyte in it. Having come to the patient, they may cause hemolysis exacerbation due to increased sensitization and complement activation.
With frequent transfusions, the formation of antioeritrocyte antibodies is possible. Such a patient, subsequent transfusion is carried out after several procedures for laundering erythrocytes with saline and testing donor blood using the Cumbas reaction.
The number of transfusions is usually assigned at least five, but depends on the severity of the patient's condition and reaction to treatment.
To stimulate the correct hemopower, a non-government (anabolic hormonal drug) by courses up to three months are used. It is possible to change the functional state of the liver.
In order to treat and prevent thrombosis, heparin is used, followed by the transition to the supporting doses of indirect anticoagulants.
To compensate for iron losses, preparations are prescribed in tablets.
Indication to the removal of the spleen can be a sharp increase, signs of heart attack. Splenectomy is rare.
Preparations of hepatoprotective action are prescribed to protect the liver. Sometimes steroid therapy helps.
The drug is administered only intravenously drip
In recent years, information has been available on the use of the medicinal preparation of echulizumab (soliris) made of monoclonal antibodies. Judging by the reports available, it blocks hemolysis, is able to confront the complement of the blood. The drug is considered the most expensive medicine in the world. His action and negative effects are not sufficiently studied.
Night hemoglobinuria has not yet have specific treatment. Even with sufficient supporting therapy, patients live about five years after the start of the disease. Prevention does not exist. Everyone should adhere to proper behavior when working and forced contact with toxic compounds.
How does paroxysmal night hemoglobinuria manifest?
Paroxysmal Night Hemoglobinuria is a severe acquired pathology of a group of hemolytic anemia. Marciaphali-Mikeli disease or Skrebinka-Markiafa disease, other names of this pathology, causes the destruction of erythrocytes in the blood. The disease is very rare, for 500 thousand people, can meet 1 person with this pathology.
In order not to worry about the development of possible complications and the consequences of pathology, you should know that it is diagnosed with paroxysmal night hemoglobinuria, symptoms and treatment of pathology.
Causes of hemoglobinuria
As mentioned above, paroxysmal night hemoglobinuria is a very rare disease, besides, pathology is most often found in people aged 20 to 40 years. Cases of the development of the disease in old age or children are also known for medical practice, but a negligible percentage falls on their share.
The cause of paroxysmal night hemoglobinuria (PNG) is considered a mutational reaction of the stem cell gene (PIG-A), which is the component of the X-chromosome in the bone marrow, in response to the impact of uncertain factors of influence. Some sources argue that the causes of the mutation of the gene are unknown.
Others argue that hemoglobinuria can develop against the background of infectious diseases, inflammation of lungs, injuries, intoxication, supercooling and burns, and even severe physical overvoltage.
But the unanimous opinion on the etiology of pathology has not yet been established.
A clear relationship of the diagnosis of the diagnosis of paroxysmal night hemoglobinuria as a symptom of associated pathologies has been revealed. Medical studies have been proven that APG is developing as the consequence of aplastic anemia and other vascular pathologies in 30% of cases.
The exactly known argument is that even one mutating cell can lead to the development of a severe form of a pathological condition. When forming erythrocytes, which is performed in the bone marrow, stem cells are divided, ripen and removed into the bloodstream. One modified gene is still divided into a pair, and they are still on a pair, etc. That is, one cell self-regulating, gradually filling blood damaged red blood cells.
The essence of damage to the erythrocytes is incomplete or absent protein membrane, which serves to protect cells from immune system. For the slightest defects of the cell, the body's immunity destroys it, as a result of which such a diagnosis is developing as hemolysis - intravascular destruction of erythrocytes, which is characterized by entering the blood of pure hemoglobin.
The same process occurs in chronic hemolytic anemia, so paroxysmal night hemoglobinuria is its analogue or, as practicing physicians, its acute acquired form. The main and only difference in these pathologies is the principle of their development.
Hemolytic anemia is a congenital pathology, hemoglobinuria - acquired. The defects of erythrocytes can be distributed on other solid elements of vascular fluid: leukocytes and platelets.
Symptoms of night hemoglobinuria
The symptoms of Markiafa-Mikeli disease depend on the causal classification of pathology. As it was found out, the disease can be independent, according to this idiopathic form of APG. Due to the development of pathology against the background of aplastic anemia, paroxysmal night hemoglobinuria acquires the shape of the syndrome. The very rare is considered an idiom form of a PNG, which arises against the background of heaping hypoplace.
Select distinct symptoms for any of the forms of the disease is impossible, as it is very variable. The course of the disease may be externally asymptomatic, in this case, it is possible to identify pathology only with laboratory diagnostics. Other patients have severe anemics syndrome.
In general, you can define a slight generalization of all possible manifestations Night hemogloburia, thus highlighting the main symptomatic picture.
- The process of hemolysis (the destruction of red blood cells and hemoglobin) occurs mainly at night (night hemoglobinuria), therefore, in the morning urination, the urine color will acquire a dark brown tint. In the day and evening, such a sign is not observed.
- Due to the quantitative decrease in the blood of the erythrocytes, anemic syndrome is observed. Its manifestations are directly connected with oxygen starvation of organs and tissues. Therefore, the patient may show headaches, dizziness, flickering black dots before eyes, general weakness, fast fatigue, attacks of angina and tachycardia.
- In case of concomitant infectious diseases, bleeding, physical activity, etc., the hemolytic crisis can develop, which is manifested by a sharp jump in the amount of hemoglobin in the vascular fluid, as well as strong malaise, fever, bone pain, may appear the jaundity of the skin and moderate splenomegaly (increasing the spleen).
- Hemoglobinuria is accompanied by a violation of the concentration of nitrogen oxide in the plasma, which, both against the background of crises, and in the severe course of pathology, causes erectile dysfunction in men.
- Due to the resulting platelet defect (blood cells responsible for blood clotting), thrombosis may occur, which are most often observed in the veins. The same process can provoke a substance that is allocated during the destruction of solid blood cells. It causes an increased coagulation of the vascular fluid, on which the tendency to thrombosis depends. Such violations may result in death.
The most distinct symptoms of paroxysmal night hemoglobinuria can be obtained during laboratory diagnostics. Studies will show the level of hemoglobin in the blood, the condition of the cells, the presence of thrombopement and leukopenia, the level of iron and other microelements, etc. For complete and accurate diagnosis of hemoglobinuria, you need a long time, since this disease can be carefully hidden under the guise of other pathologies.
Therefore, the most rational way of timely identification of marciaphali-mixeli disease will be a regular prophylactic examination.
Treatment of paroxysmal night hemoglobinuria
The period of detection of paroxysmal night hemoglobinuria determines the necessary medical methods and establishes the forecast of the outcome of pathology, which in most cases unfavorable. This is due to the lack of a specific reason for the development and inability to eliminate it. Therefore, there is no specific PNG treatment technique.
All therapeutic measures are aimed at eliminating symptomatic manifestations. The only one in an effective way Full getting rid of mutated cells is a red bone marrow transplant (place where blood cells are formed).
With the development of a hemolytic crision, the acute form of hemolysis, the patient is prescribed a multiple transfusion of the erythrocyty mass. Such transfusions can be 5 or more. The number of procedures and their frequency is determined by repeated analyzes and is carried out at the next reproduction of defective red blood cells.
In rare cases, the removal of the spleen is carried out. Symptoms leading to splenectomy are a sharp increase in the body and the manifestation of the development of the infarction state.
The remaining therapeutic measures are made in the reception of different group drugs that facilitate the course of pathology. The main drugs are the preparations of groups of steroid hormones, cytostatics, as well as iron and folic acid preparations.
Nero
The most frequent in the appointment of doctors to combat the symptomatic manifestation of paroxysmal night hemoglobinuria is the drug NerCol. This is the hormonal drug of the group of anabolic steroids. The effect of the drug is directed:
- to stimulate the synthesis of protein in the organism of the patient, which is not enough in the defective erythrocyte membrane;
- has a beneficial effect on nitrogen exchange;
- delays the withdrawal of potassium, sulfur and phosphorus, which are necessary for normal protein synthesis;
- provocates the enhanced fixation of calcium in the bones.
After receiving this drug The patient feels an increase in appetite, intensive increase in muscle mass, accelerating the calcination of bones, and is also much better general state organism.
The use of the drug begins with 10 g, gradually increasing to 30 g of 1-2 reception per day. For children, the dose of the drug is 1 tablet every other day, with severe form daily. The course of therapy is nervous ranging from 2 to 3 months.
After the end of the use of the drug, many patients have increased hemolysis.
The use of the necocol can be carried out strictly by appointing the attending physician.
Heparin
Heparin is a direct anticoagulant - a means for braking blood clotting. With paroxysmal night hemoglobinuria, it is prescribed to prevent thrombosis that complicate the course of the disease.
The dosage and reception frequency is fully individualized, depending on the complexity of the pathology and risk of blood clots in vessels.
At the end of the course heparin, the doctor prescribes an indirect anticoagulant to maintain normal level coagulation.
Ekulizumab
Echulizumab is a drug that consists of humanized monocanal antibodies. The principle of the drug is to stop intravascular hemolysis and direct confrontation of the blood compliment. As a result, the natural destruction of defective erythrocytes of the body's immune system stops.
This drug is the most expensive medicine in the world. Its mechanism of action and development possible consequences Applications are not studied enough.
Iron and Folic Acid Preparations
In case of violations in the work of the red bone marrow, iron and folic acid deficiency arises, which are necessary for normal blood formation. The drug therapy of the PNG includes the reception of drugs of these trace elements, to compensate for pathological losses.
The dosage and method of receiving the drug is determined by the attending physician. Most often soribifer, trifeferron, ferretab, phenyuls, etc. are prescribed. The composition of these drugs includes a complex of trace elements necessary for the normal creation of solid blood particles in the red bone marrow.
Support liver
Enhanced therapy in the fight against paroxysmal night hemoglobinuria strongly affects the liver. In the absence of supporting therapy for the liver, it can simply refuse. Therefore, it is important to take the preparations of hepatoprotective action. These can be such drugs:
In addition, there are a number of products that contribute to the restoration of liver cells. These include pumpkin, kuraga, laminaria, olive oil, dairy products and much more. The main thing in the moments of the weakness of the liver do not exacerbate its harmful food.
After identifying the disease, doctors give inaccurate forecasts. Statistics say that after establishing a diagnosis, the patient can live on supporting therapy for about 5 years.
Due to the unknown origin of the disease and inaccuracies in the causes of its development, the paroxysmal night hemoglobinuria is impossible to propagate.
conclusions
Marciaphali-Mikeli disease or paroxysmal night hemoglobinuria is a severe disease, which even in intensive therapy leads to a fatal outcome. The only possible rehabilitation is the transplantation of a red bone marrow, which forms blood cells. In addition, pathology entails the development of concomitant diseases that are no less dangerous for the state of the patient.
Therefore, doctors unanimously declare that the best method to propagate any pathology is a regular passage of a full medical examination. It is possible if the disease is only at the formation stage can be permanently removed. With such serious diseases, the main question is time. You should take care of yourself and your own body.
Paroxysmal Night Hemoglobinuria (PNG)
The reasons:
Causes of disease are associated with intravascular destruction of erythrocytes, defective in a significant part. Along with the pathological population of erythrocytes, some of the normal cells that have a normal life lifetime are also preserved. Disruptions in the structure of granulocytes and platelets are found. The disease is not hereditary, but any external factors provoking the formation of a defective population of cells, which is a clone, i.e. The offspring of one initially modified cell is not known.
There is an increased complementaryness of pathological erythrocytes with APG. Perhaps this is the basis for provocating a hemolytic crisis with a transfusion of fresh blood, which contains factors that activate complement. The transfusion of blood stored for more than a week, hemolysis does not provoke.
Symptoms of paroxysmal night hemoglobinuria:
The disease develops slowly: signs of moderate anemia, weakness, fatigue, heartbeat under load, abdominal pain, often associated with thrombosis of mesenterial vessels appear. The skin and mucous membranes are pale yellow, grayish in connection with anemia and hemosideric deposition. Characteristic signs of intravascular hemolysis.
The morphology of erythrocytes does not have characteristic features. In the bone marrow there is a hyperplasia of a red sprout, but in Trepanate there is a slight increase in the bone marrow cell, which, as the disease, it can become hypoplastic.
Diagnosis:
The diagnosis is established on the basis of signs of intravascular hemolysis (anemia, small reticulocyte, hemosiderer in the urine). Specify the diagnosis with special studies (positive sucrose sample, hem test, negative Cumbac sample).
In contrast to the APG, there is no leukopenia and thrombocytopenia, usually a good effect gives prednisolone. DEVERATE FROM APLASTIC ARMIA PHB Allows the pattern of bone marrow: in Aplasia, the trepanat is characterized by the predominance of fat, during hemolysis - cell hyperplasia, but in rare CHANS, the pattern of bone marrow hypoplasia can develop, although hemosidemin is constantly detected in the urine, and reticulocytosis in the blood.
Treatment of paroxysmal night hemoglobinuria:
Treatment in the absence of pronounced anemia is not carried out. Heavy anemic syndrome requires transfusion of red blood cells; The best results give the transfusion of washed or weathered within 7-10 days of erythrocytes. In the heap formation hypoplace, anabolic steroids are shown: Nerochol - 10-20 mg per day or retabilic - 50 mg intramuscularly within 2-3 weeks.
Discussions
PNG (Paroxysmal Night Hemoglobinuria) PNG
12 messages
APG is a complex disease that is manifested by non-specific and unpredictable signs and symptoms, often coinciding with such other diseases. In addition, each PNG patient manifests itself in different ways. If you have a PNG, then some of the erythrocytes or all erythrocytes of your blood are deprived of an important protective protein. Without this protein, red blood cells can be collapped under the influence of one of the components of the immune system of your body, called the complement system. Even if you do not feel it, hemolysis is constantly, hidden and can pose a threat to your life. As in the case of other chronic diseases, such as diabetes or arterial hypertension, Lack of treating APG may result in serious health violations. Typical symptoms associated with APG include abdominal pain, difficulty swallowing, anemia, shortness of breath and fatigue. More serious complications include the formation of blood clots, renal failure and damage to vital organs.
Patients of APG may have different manifestations of the disease, which can be unpredied from time to time (for example, under stress) or improve. However, all patients with PNG observes chronic hemolysis.
PAG patients may also have other diseases that affect the function of bone marrow, for example aplastic anemia or myelodsplastic syndrome. In contrast to the PHG, in which the red blood cells are destroyed, the production of blood cells can decrease with these diseases, which further complicates the CNG current. If you are diagnosed with PNG simultaneously with aplastic anemia or myelodic syndrome, discuss with your doctor all possible effective treatment options for you have.
With APG, red blood cells are deprived of an important protein
2. Activities of the Complement system
Without this protein, part of the red blood cells can be destroyed under the influence of the complement system, one of the protective systems of the body.
3. Development (hemolysis) erythrocytes with APG
With the PNG, the destruction of red blood cells occurs, as a result of which the toxic decay products fall into the surrounding plasma (liquid blood component of yellow color).
Hemolysis is a medical term meaning "destruction of red blood cells." The intensity of hemolysis is assessed by the results of determining the activity of LDH (lactate dehydrogenase - the enzyme contained in the erythrocytes). Increased LDH activity indicates excessive hemolysis. W. healthy people Minor hemolysis is a natural constant process. However, patients with PNG observes excessive hemolysis due to the lack of protective protein on the surface of the part or all red blood cells. Such excessive hemolysis is accompanied by the release of erythrocyte toxic content into the blood, which can eventually lead to the emergence of the majority of symptoms associated with APG, as well as to damage the important organs of your body. If you are sick pnts, hemolysis is constantly, regardless of whether you feel good or with you exacerbating (paroxysm) of the disease, such as, for example, during stress or in infection. Excessive and prolonged hemolysis is the main cause of serious human health violations.
In the destruction of blood cells, their toxic content falls into the blood and can accumulate there, causing, thereby harm to health that may appear unexpectedly and at any time. Such violations may include renal failure and the formation of dangerous thrombans, which can lead to violations of such vital organs, such as liver, brain and lungs.
Hemolysis also affects your well-being. Many PNG patients note that the unpredictability of the appearance and severity of the symptoms of the disease adversely affects the quality of their lives. Reducing chronic hemolysis is considered by doctors as an essential target in the treatment of APG.
The drug is antibodies that block the C5 component of the Complement system. Application experience showed an increase in survival, a decrease in hemolysis and thrombosis, improving the quality of life.
The disease most often begins. Patients complain of weakness, malaise, dizziness. Sometimes the subcomfation is scler. Often the first are complaints about headache, pain in the stomach of various localization. The tendency to elevated thrombosis causes the patient to consult a doctor. Hemoglobinuria is quite rarely the first symptom of the disease and in some PNG patients may not be absent. In some cases, it appears for the first time in 2-3 years and even 10 years after the onset of the disease.
One of the characteristic signs of APG - attacks of abdominal pain. Localization of it may be the most different. Outside the crisis period, the abdominal pain is usually not observed. Often it is joined by vomiting. Most likely, pain in the abdomen in patients with PNG is associated with thrombosis of mesenteric vessels.
Thrombosis of peripheral vessels (most often the veins of the upper and lower extremities, less often - kidney vessels) also characteristic symptom Paroxysmal night hemoglobinuria. In 12% of PNG patients, thrombophlebitis is observed. Thrombotic complications are the most frequent cause death with a given disease.
With an objective study of the patient, attention is most often drawn to the pallor with a small jaundice tint. Often there is an endlessness of the face, sometimes excessive fullness. It is possible a small increase in the spleen and liver, although it is not typical for the APG.
For paroxysmal night hemoglobinuria, signs of intravascular hemolysis are characterized, the most important of which is the increase in the free hemoglobin of plasma. This feature is periodically marked by almost all PNG patients. However, the degree of increasing the free hemoglobin of plasma varies and depends on what period of the disease was conducted. During the crisis period, this figure increases significantly, there is also an increase in the amount of plasma metalbumin.
The level of free hemoglobin plasma depends on the degree of hemolysis at the moment, the content of haptoglobin, the degree of filtering hemoglobin with urine and on the destruction of the hemoglobin complex - Gaptoglobin. In the case of a small degree of hemolysis, the level of free hemoglobin plasma will be insufficient to filter it through the renal filter. Therefore, hemoglobinuria is not a mandatory symptom of the disease. When passing through the tubes of nephrons, hemoglobin is partially destroyed and deposited in the epithelium of the tubules. This is the cause of hemosidery with urine.
Hemosiderine stands out with urine at the overwhelming majority of patients with paroxysmal night hemoglobinuria. Sometimes hemosyrinuria does not appear immediately. This is an important, but not specific for APG a sign of the disease.
Laboratory Indicators for Paroxysmal Night Hemoglobinuria
The number of erythrocytes in patients with PNG decreases, respectively, the level of reduction of hemoglobin. The color indicator remains close to one. In cases where the patient loses a significant amount of iron with the urine in the form of hemosiderin and hemoglobin, the iron level is gradually decreasing. Low color indicator is observed about half of the patients. Some of them have increased the level of hemoglobin P, especially during the exacerbation.
In a large part of the patients, the content of reticulocytes is increased, but relatively low (2-4%). The number of leukocytes with APG in most cases is reduced. In many patients, it is 1.5-3 g in 1 liter, but sometimes decreases to 0.7-0.8 g in 1 liter. Lakeing, as a rule, is observed by reducing the number of neutrophilic granulocytes. Sometimes the content of leukocytes with PNG is normal or elevated - to 10-11 g in 1 liter.
Paroxysmal night hemoglobinuria, also known as the Disease of Strejbelga-Markiafa, Markiafa-Mikeli disease, is a rare disease, progressive blood pathology, threatening the patient's life. It is one of the species of acquired hemolytic anemia due to violations of the structure of the erythrocyte membranes. Defective cells are susceptible to premature decay (hemolysis) flowing inside the vessels. The disease has a genetic nature, but is not considered inheritable.
Night paroxysmal hemoglobinuria
Epidemiology
Meeting frequency - 2 cases per 1 million people. The incidence of 1.3 cases per million people during the year. It is mostly manifested in persons in an eating, the dependence of the incidence of gender and the race is not detected. A single cases of the disease in children and adolescents are known.
Important: the average age of the detection of illness is 35 years.
What is Night Paroxysmal Hemoglobinuria
Causes of the disease
The causes and factors of the risk of developing the disease are unknown. It has been established that pathology is due to the mutation of the Pig-A gene, located in the short shoulder X-chromosome. The mutagenic factor is currently not set. In 30% of cases of incidence of night paroxysmal hemoglobinuria, communication with another blood disease - aplastic anemia is traced.
Education, development and ripening of blood cells (hematopoEEZ) flows in the red bone marrow. All specialized blood cells are formed from the so-called stem, non-specialized, cells that have preserved the ability to divide. Formed as a result of consecutive divisions and transformations, ripe blood cells overlook the bloodstream.
The Mutation of the Pig-A gene leads to the development of PNG even in a single cell. The damage of the gene changes and the activity of cells in the processes of maintaining the volume of bone marrow, mutant cells multiply more active than normal. In the hematopoietic fabric, the population of cells producing defective uniform elements is quite quickly formed. At the same time, the mutant clone does not apply to malignant formations and can disappear spontaneously. The most active substitution of normal bone marrow cells mutant occurs in the processes of restoration of bone marrow tissue after significant lesions caused, in particular, aplastic anemia.
Characteristic signs of night paroxysmal hemoglobinuria
Damage to the Pig-A gene leads to violations of the synthesis of signal proteins that protect the organism cells from the effects of the complement system. Complement system - specific blood plasma proteins that provide overall immune protection. These proteins are associated with damaged erythrocytes and melted them, and the hemoglobin released is mixed with blood plasma.
Classification
Based on the available data on the causes and peculiarities of pathological changes, several forms of paroxysmal night hemoglobinuria are distinguished:
- Subclinical.
- Classic.
- Tied with impaired hemopoiesis.
Subclinical form of the disease is often preceded by aplastic anemia. Clinical manifestations There is no pathology, however the presence of a small amount of defective blood cells is detected only with laboratory studies.
Night Paroxysmal Hemoglobinuria Clinic
On a note. There is an opinion that the PNG is a more complex disease, the first stage of which is aplastic anemia.
The classic form proceeds with the manifestations of typical symptoms, in the blood of the patient there is a population of defective erythrocytes, platelets and certain types of leukocytes. Laboratory methods Studies confirm the intravascular destruction of pathologically modified cells, hemopoix disorders are not detected.
After transferring diseases that lead to insufficiency of blood formation develops a third form of pathology. Pronounced clinical picture And the intravascular lysis of red blood cells develop against the background of bone marrow lesions.
There is an alternative classification according to which it is distinguished:
- Actually pnts, idiopathic.
- Developing as accompanying syndrome with other pathologies.
- Developing as a consequence of bone marrow hypoplasia.
Night paroxysmal hemoglobinuria clinic. Part 2
The severity of the course of the disease in different cases is not always interrelated with the number of defective erythrocytes. Describes both cases of subclinical flow when the content of modified cells approaching 90% and extremely heavy, with a replacement of 10% of the normal population.
Development of the disease
At the moment, it is known that three types of erythrocytes with different sensitivity to the destruction of the complement system may be present in the blood of patients with paroxysmal night hemoglobinuria. In addition to normal cells, erythrocytes are circulated in the bloodstream, the sensitivity of which is at times exceed normal. In the blood of patients with diagnosed Markiafa-Mikeke disease, cells were found, the sensitivity of which to complement is higher than the norm in 3-5 IARS.
What does Night Paroxysmal Hemoglobinuria
Pathological changes affect other uniform elements, namely platelets and granulocytes. In the midst of the disease in patients, pancytopenia is detected - the insufficient content of blood cells of different types.
The severity of the manifestation of the disease depends on the relationship between populations of healthy and defective blood cells. The maximum content of erythrocytes, super-sensitive to complement-dependent hemolysis is achieved within 2-3 years from the moment of mutation. At this time, the first typical symptoms of the disease appear.
Pathology is usually developing gradually, the sharp crisis began is rarely observed. Exacerbations manifest on the background of menstruation, strong stress, sharp viral diseases, surgical intervention, treatment with some drugs (in particular, iron-containing). Sometimes the disease is exacerbated by eating some products or without obvious reasons.
Paroxysmal night hemoglobinuria
There is data on manifestations of Markiafa-Mikeli disease due to irradiation.
The dissolution of blood cells to one degree or another in patients with a paroxysmal night hemoglobinurion installed constantly. Periods of moderate flow are mixed with hemolytic crises, mass destruction of red blood cells, which leads to a sharp deterioration in the patient's condition.
Outside the crisis of patients, the manifestations of moderate general hypoxia are worried, such as shortness of breath, attacks of arrhythmias, general weakness, worsen the tolerability of physical exertion. During the crisis, abdominal pains, localized mainly in the field of navel, in the lower back are manifested. Water stained in black, the darkest portion is the morning. The reasons for this phenomenon are not yet established. The PHG develops minor pastosity of the face, the yellowness of the skin and the scler is noticeable.
On a note! A typical symptom of the disease - urine dyeing. Approximately half of the known cases of the disease are not manifested.
Changing the urine color from the norm to pathology in paroxysmal night hemoglobinuria
In periods between cries, patients may be observed:
- anemia;
- tendency to thrombosis;
- increase liver;
- manifestations of myocardiodistrophy;
- next to infectious infection.
In the destruction of blood cells, substances are released, increasing the coagulation, which causes thrombosis. The formation of blood clots in the vessels of the liver, kidneys, the defeats are also susceptible to coronary and cerebral vessels, which can lead to a fatal outcome. Thrombosis localized in the vessels of the liver leads to an increase in the size of the organ. Disturbance of intrahepter blood flow entails dystrophic changes in tissues. When blocking the system of portal veins or veins of the spleen develops splenomegaly. Violations of nitrogen exchange are accompanied by impaired functions of smooth muscles, some patients complain about difficulty swallowing, esophageal spasms, men have erectile dysfunction.
Important! Thrombotic complications in the PHG predominantly affect veins, arterial thrombosis rarely develop.
Video - Paroxysmal Night Hemoglobinuria
The mechanisms for the development of complications of APG
Hemolytic crisis is manifested by the following symptoms:
- acute abdomen pain caused by multiple thrombosis of small mesenterical veins;
- enhancing the jaggility;
- pain in the lower back;
- decrease in blood pressure;
- increased body temperature;
- staining urine in black or dark brown color.
In rare cases, the hemolytic kidney develops, a specific transient form of renal failure, accompanied by acute Anuria. Due to the violation of the separation function in the blood, nitrogen-containing organic compounds are accumulated, which are finite products of protein decay, azotemia develops. After the failure of the patient from the crisis, the content of uniform elements in the blood is gradually restored, the jaggility and manifestations of anemia are partially faded.
The most common variant of the course of the disease is critical, intermitted by periods of a stable satisfactory state. In some patients, periods between crises are very short, insufficient to restore blood composition. Such patients develop steady anemia. There is also a variant of the current with acute start and frequent crises. Over time, crises are becoming less. In particularly severe cases, a fatal outcome is possible, which cause acute renal failure or thrombosis of the vessels feeding the heart or brain.
Important! Daily laws in the development of hemolytic crises were not revealed.
In rare cases, the disease may have a long-term calm current, isolated cases of recovery.
Diagnostics
Laboratory diagnostics of night paroxysmal hemoglobinuria
On the early stages Diagnosis disease is difficult due to the manifestation of scattered nonspecific symptoms. For diagnosis, sometimes several months of observations are sometimes required. A classic symptom is a specific urine staining - manifests itself during crises and not in all patients. The grounds for suspicion of Markiafa-Mikeli disease are:
- iron deficiency of unknown etiology;
- thrombosis, headaches, attacks of lower back pain and belly without visible reasons;
- hemolytic anemia of unexplained origin;
- the melting of blood cells accompanied by pancytopenia;
- hemolytic complications associated with the transfusion of fresh donor blood.
In the process of diagnosis, it is important to establish the fact of chronic intravascular decay of the erythrocytes and identify specific serological signs of APG.
In a complex of research, with suspected night paroxysmal hemoglobinuria, in addition to general urinary tests and blood are carried out:
- determination of the content of hemoglobin and haptoglobin in the blood;
- immunophenotyping by flow cytometry to identify populations of defective cells;
- serological samples, in particular, Cumbac sample.
Diagnostic tests at night paroxysmal hemoglobinuria
Needless differential diagnosis with hemoglobinuria and anemia of other etiology, in particular, to exclude autoimmune hemolytic anemia. General symptoms are anemia, jaggility, increasing the content of bilirubin in the blood. The increase in the liver or \\ and spleen is not observed in all patients