Bilyiary liver cirrhosis Clinical recommendations. Modern consensus on the diagnosis and treatment of primary biliary cirrhosis and primary sclerosing cholangitis. Diagnosis and therapy
Citation:Pyymova S.D. Primary biliary cirrhosis // RMW. 2002. №2. P. 57.
Primary biliary cirrhosis (PBC) is a chronic granulomatous destructive inflammatory disease of the interdollak and septal bile ducts of autoimmune nature, leading to the development of a long cholestasis, and in later stages to the formation of a cirrhosis.
Etiology and pathogenesis
Etiology PBC is unknown. A certain role is played by genetic factors. Cases of family diseases are described, but their frequency is small - 1-7%.
The leading value in the pathogenesis of the PBC has autoimmune cellular reactions. An autoimmune diseases of the liver are characterized by the presence of specific autoantibodies. For PBC, it is characteristic of the presence of antimitochondrial antibodies (AMA), specific for complexes of dehydrogenase 2-oxocoslotes located on the inner membrane of mitochondria. The most common (95-100%) at the PBC is detected by autoantoantibodes to E2 component of the pyruvate dehydrogenase complex (RDS-E2).
For a long time it was believed that the presence of AMA is only an accompanying sign, but after Gershwin and Mackay discovered autoantigen, the studied studies revealed the specificity of the AMA's action, and their role in the pathogenesis of the disease was disclosed. These antibodies suppress the activity of the RDS-E2 acting in the role of an immunodominant target. AMA is IgG3 and IgM, detected in serum and bile of patients. The corresponding epitons of B cells are described. The correlations between the number of AMA and the disease stage were not detected, but the dependence between the activity of the process and the level of PBC-specific B-cells in the blood serum is shown.
The central target for the development of inflammatory response and immune response are bile ducts. AMA is binding to the apicial membrane of epithelial cells of bile ducts, on the surface of which proteins of the main histocompatibility complex (MNS) of class II are located. It can be assumed that the pathological expression of autoantigen occurs earlier than an immune response with expression on the surface of class II proteins cells is formed. Further expression occurs in the later stages of the development of the disease, the presence of activated T-cells is associated with the nec-inflammatory process in bile ducts. It is important to note that the adhesion molecules that enhance the immune response are detected on the cells of the biliary epithelium and on lymphocytes.
T-lymphocytes play the main role in direct damage to intrahepken bile ducts. In the liver and peripheral blood of patients, CD4-positive RDS-E2-specific T-helpers are found - both TX1 and TX2-populations. It is evidence that in the liver of patients with PBC TX1 cells prevail, they are stimulated by a cellular immune response by means of IL-2 products and info.
The answer to the question of how the RDS-E2, which is the peptides of the organism itself, can cause an immune response, gives the theory of molecular mimicry.
The main mechanism of the death of the bile epithelium cells is the apoptosis, which is carried out both TX1, carrier-ligand, and secreted by this cell subpopulation of cytokines.
Morphological characteristic
Currently, a classification was adopted, according to which 4 histological stages of PBC are distinguished: chronic non-resident destructive cholangitis - the duktal stage; Proliferation of bile ducts and perideuccotal fibrosis - Duktool Stage; fibrosis of stroma in the presence of inflammatory infiltration of the liver parenchyma; cirrhosis of the liver.
Chronic non-resident destructive cholangitis (1st stage) is characterized by inflammation and destruction of predominantly intercroid and septal bile ducts. Extended portal paths are infiltrated with lymphocytes, plasma cells, macrophages and eosinophilic leukocytes. Among the cells of portal paths are formed lymphoid follicles. The infiltrate of portal paths does not apply to a parenchyma, separate lymphocytes or cell groups can penetrate into the slices. Infiltrates are found in the walls of some intra-robber bile ducts.
The integrity of the basal membrane of the affected bile ducts is broken.
Often near the affected bile ducts show granuloma - granulomatous cholangitis. Granulomas are built from epithelioid and giant multi-core cells and in most cases are well distinguishable in preparations.
Histological signs of cholestasis in this stage are usually not detected.
Proliferation of cholangiol and perideoscotal fibrosis (2nd stage). In portal paths, along with lymphoplasmocyte infiltration and destructive bile ducts, the foci of the proliferation of biliary epithelium appear. Proliferating cholangiols with cells of infiltrate are distributed to the periportal departments of the poles. The number of interdollastic and septal bile ducts as they are destructive decreases. A characteristic diagnostic sign of the PBC is appearing - "empty" portal paths, inflammatory infiltrates that do not contain bile ducts.
Fibrosis of stroma in the presence of inflammatory infiltration of the liver parenchyma (3rd stage) is characterized by the appearance of connecting lights that are separated from the portal paths and interconnecting adjacent paths (portal septiles) and central veins with portal paths (portocentral septa). In them, the inflammatory infiltration applies to proliferating bile ducts, the proliferation of the ducts is reduced. Progresses the reduction of interdolk and septal bile ducts. This leads to strengthening cholestasis. Muchly increases the copper content in liver biopsy.
Cellular infiltration of parenchyma and hepatocyte necrosis increases, fibrosis increases in portal paths, monolobular false slices are formed.
Cirrhosis of the liver (4th stage) is characterized by all signs of monolobular cirrhosis.
Clinical picture
The disease is predominantly in women, more often over the age of 35. Distinctive feature PBC is a relatively rare morbidity of men (10-15% in the total incidence of PBC).
Skin itch is the most characteristic initial symptom PBC, observed in most patients. The skin itch is combined with yellow skin and scool staining, but often it precedes jaundice, sometimes in a few months and even years. A number of patients observed by us for 2-6 years developed only the light yellowness of the scool without staining of the skin.
The jaundice of the cholestatic type, slowly increasing, is detected as an early symptom of the disease in less than half of the patients. Jaundice appearing at the time of establishing a diagnosis and rapidly increasing can be considered as a prognostically unfavorable symptom indicating the rapid progression of the disease.
Xantellasma in the early stages are determined in 20-30% of patients. Their formation directly depends on the level and duration of hypercholesterolemia. Extraked signs - "liver" palms, vascular asterisks are only in individual patients; They are always single. Most of the observed men revealed gynecomastia.
Hepatomegaly is usually minor, detected in most patients. Splenomegaly is observed in less than half of the patients, does not combine with hyperplane phenomena. In the early stages, the demineralization of the bones appears pain in the lower back, ribs, joints.
The initial signs of the disease can serve as nonspecific symptoms as pain in the field of right hypochondrium, in some cases with fever; Elevated ego; pain in the joints and muscles, as well as dyspepsic, skin syndromes, vasculitis, sclerodermia. In 20% of patients on initial stages The disease can occur without clinical symptoms, while the SCF is often increased, AMA is always detected in the titer 1:40 and higher, changes are detected in the biopsy of the liver, characteristic of PBC.
The deployed PBC stages are characterized by a progressive deterioration in the state of patients, an increase in jaundice, sometimes increasing the temperature to subfebrile, and then febrile digits, exhaustion (up to cachexia) due to impaired absorption in the intestine. The skin of the skin in the terminal stage of the disease in a number of patients weakens, and with progressive hepatic cellular insufficiency disappears.
With the progression of cholestasis, steato seater, osteoporosis, and then osteomalacia, xerofthalmia and hemorrhagic syndrome. The fragility of the vertebral bodies, kyphosis and pathological fractures appear. Signs of portal hypertension are developing, in particular, veins of the esophagus and stomach are evil. Patients die under the phenomena of hepatic cellular insufficiency, which can provoke complications of biliary cirrhosis: bone fractures, portal hypertension, ulcerative bleeding.
For later complications, PBC should include the development of cholangiocarcinoma, significantly more often observed in men than in women. It is also possible to form stones in the bustling bubble.
System manifestations
For biliary cirrhosis, the systemic lesions is natural, the most brightly manifest changes in the exocrine glands: tear, salivary, pancreas, as well as kidney (tubul-interstitial jade, glomerulonephritis) and vascular (vasculitis) of various organs.
SHEGREEN syndrome at a targeted examination is detected in 70-100% of patients with biliary cirrhosis. The involvement of the lacrimal and salivary glands during the SHEGREEN syndrome is most often clinically manifested by dry keratoconjunctivitis, xerostomy, decrease in tear when the broader sample, recurrent pairotite and dry skin.
The pulmonary syndrome, observed in patients with biliary cirrhosis, rather, x-ray, than clinical, and is characterized by a pattern of diffuse pneumosclerosis with deformation of the pulmonary pattern due to additional heavy, petrose and cellular tissues for interstitial type and fibrosising alveolitis.
Accompanying illnesses
PBC is combined with other chronic diseases, mainly autoimmune nature - sclerodermia, rheumatoid arthritis, thyaredite Hashimoto, Miastenia, celiac and adult, transverse myelitis. Combined autoimmune disorders in women naturally occur more often than men. The frequency of insulin-dependent diabetes mellitus in men is higher than that of women.
Sclerodermia. The combination of PBC with sclerodermia, according to various authors, ranges from 3 to 18%. In some cases, clinical manifestations of sclerodermia correspond to the Crest syndrome (calcine, Reyno syndrome, dysfunction of the esophagus, sclerodcticity, teleangioectasis). The leather, mucous membranes, joints, musculatura are involved in the pathological process. With a combination of sclerodermia and PBC, clinically pronounced damage internal organs are usually absent, which determines the benign course of the disease. In the blood, antinuclear antibodies and rheumatoid factor usually determine.
Systemic red lupus. Characteristic of the diversity and severity of manifestations: skin, articular, muscular syndromes, lymphadenopathy, polyperozit, kidney damage, lungs, hearts, nervous system, hemocytogen. The progression of diseases usually leads to the death of patients in 3-7 years after the appearance of the first symptoms. LE cells and antibodies to native DNA are found in the blood.
Rheumatoid arthritis. The frequency of rheumatoid arthritis in patients with PBC is up to 10%. They are affected mainly interfalane, rays-up, knee, ankle joints. The main symptoms are the pain and edema of the joints, the violation of mobility in them, generalized lymphadenopathy, musculature atrophy in the area of \u200b\u200baffected joints. When radiological examination, osteoporosis of the bones of the joints of the joints involved, the narrowing of the intermediate slots, the uzura of the articular surfaces. The rheumatoid factor is determined in serum, articular fluid, and also by the immunofluorescence reaction in the field of lymphoid infiltration of the synovial shell.
The defeat of the thyroid gland, according to various authors, is observed in 18-32% of cases at PBC. The overwhelming majority of patients have clinical picture Hypothyroidism. We observed a combination of thyaredite Hashimoto with PBC in 3 women aged 48-52 years. A significant increase in the thyroid gland, diffuse and noded, appeared in 2 patients against the background of cirrhosis, and in one - 1 year before the development of cholestasis. In the blood, they are determined mainly anti-hydrogen and antimicrosal antibodies.
Other autoimmune diseases can also be combined with PBC: autoimmune thrombocytopenia, fibrusing alveolitis, pernicious anemia, sarcoidosis, renal tubular acidosis. Skin lesions with presumably immune pathogenesis with PBC are most often associated red flat deprived.
With the development of the immunodeficiency state, especially in cases of immunosuppressive therapy, they associate a high frequency of malignant tumors of extrahepatic localization in patients with PBC. Breast cancer reveal in women with PBC 4.4 times more often than in the overall population.
Laboratory data
Already in the early stages, it is characterized by an increase in the activity of cholestasis enzymes: paragraphs, leucinopeptids, G-glutorattranspendase. Increasing the level of serum bilirubin by 1.5-3.5 times compared with the norm is observed later and slowly increasing. The concentration of bile acids and the copper content in the blood serum increases, and the iron level decreases. Already at the beginning of the disease, pronounced hyperlipidemia with an increase in cholesterol concentration, b-lipoproteins, phospholipids and non-inteterified fatty acids. Values \u200b\u200bof aminotransferase serum are increased by 2-3 times, their activity correlates with histological data.
Of particular importance in the diagnosis of PBC is AMA. Currently, antibodies are known to 9 antigens of the internal and external mitochondrial membrane. Of these, the PBC is associated with anti-m2, -m4, -m8, -m9. The remaining antibodies are associated with other diseases: anti-M1 with syphilis, anti-M5 - with diseases of the connective tissue, anti-m3 - with medicinal hepatitis, anti-M7 - with myocarditis. Antibodies to an antigen of the inner membrane Mitochondria M2 detect almost in all cases of PBC and consider pathogenomous for this disease. The AMA to the M4 is detected with the disease with the features of both PBC and autoimmune hepatitis (overlap-syndrome), to M8 - with a fast-moving form of PBC, to M9 - in the early stages of PBC.
The titer of antimitochondrial antibodies often correlates with the activity of PBC. AMA may be discovered on a preclinical stage and do not disappear throughout the entire period of the disease.
Diagnosis
It is necessary to take into account the floor, age, heredity, it should be especially emphasized that in 1/3 cases the disease is diagnosed in women over 60 years. The most important clinical symptom - skin itch. In the early stages of the disease, the activity of cholestasis enzymes is increased, the ETE acceleration is noted. Antimitochondrial class M2 antibodies is a specific and valuable diagnostic test. With ultrasound, CT detects unchanged extratect bile ducts.
Confirms the diagnosis of the histological examination of the liver bioptate, with the help of which ungown destructive cholangitis is detected in the early stages of the disease, later - the formation of biliary liver cirrhosis.
Diagnostic Criteria PBC:
1. Intense skin itching, clinical suspicion based on the presence of extrahepatic manifestations (dry syndrome, rheumatoid arthritis, etc.).
2. Increasing the level of cholestasis enzymes 2-3 times compared with the norm.
3. Normal extratect bile moves when ultrasound.
4. Detection of antimicochondrial antibodies in the title above 1:40.
5. Increased IGM levels in serum.
6. Characteristic changes in the liver point.
The diagnosis of PBC is posed in the presence of the 4th and 6th criteria or 3-4 of these features.
Differential diagnosis
PBC must be distinguished from a number of diseases accompanied by hepatobiliary obstruction or cholestasis.
The most important diseases with which the PBC are differentiated in adults:
. Obstruction of extrahepatic biliary moves: Conductors, strictures, tumors;
. primary sclerosing cholangitis;
. Karcinoma of intrahepatic bilia tract;
. autoimmune hepatitis;
. cholestasis caused by medicines;
. chronic viral hepatitis C;
. Sarcoidosis.
In the children's and youthful age PBC differentiate from:
- hypoplasia of intrahepatic bile ducts,
- cholangiodysplasia (congenital liver fibrosis),
- biliary cirrhosis with fiberglass.
The differentiation of the PBC from the obstruction of extrahepatic bile moves is most important, since often patients with PBC are subject to unjustified laparotomy about the proposed backing jaundice, and the correct diagnosis is made only after the liver biopsy.
For the differential diagnosis of the PBC with the obstruction of extrahepatic bile strokes, primary sclerosing cholangitis, hypoplasia of intrahepatic bile ducts, congenital liver fibrosis, along with the study of antimitochondrial antibodies, direct visualization of biliary wood is needed (endoscopic sonography, retrograde endoscopic or percutaneous christapping cholangiography).
Differential diagnosis in the early stages of PBC with autoimmune hepatitis in the absence of a clear histological picture in 15% of cases cause significant difficulties. However, the detection of immunological phenomena such as antimitochondrial antibodies of class M2, the prevalence of IGM serum, and in the liver biopsy, prevailing the lesion of bile ducts over changes in the liver parenchyma, the destruction of interdollak and septal ducts, makes it possible to diagnose PBC. Such features of the disease as the high activity of aminotransferase, the detection of antibodies to smooth muscles can serve as guidelines for detecting autoimmune hepatitis.
In some cases, PBC has to distinguish with chronic cholestasis caused by drugs. Unlike PBC, drug cholestatic hepatitis proceeds with a less pronounced destruction of interdolkovoy biliary strokes and nonresut cell infiltration of portal paths; Antimitochondrial antibodies are absent; Cancellation of drugs most often leads to the reverse development of the process.
The greatest difficulties arise in the delimitation of PBC with medicinal cholestasis, accompanied by autoimmunization markers. In the liver biopsy in these cases, epithelioid-cell and giant granulomas are often detected, which differ from PBC with a large number of eosinophilic leukocytes. After the abolition of drugs, the granulomatous reaction is replaced by fibrosis.
The forecast depends on the stage of the disease. Since the appearance of the first clinical signs, the PBC is characterized by a gradual, over 12-20 years the progression of the pathological process. Among the prognostic models, the MEIO clinic model, taking into account the age, level of bilirubin, serum albumin, prothrombin time, and the presence of ascites are most often used. The terminal stage is characterized by increasing liver failure, the appearance of ascites, hepatorenal syndrome, encephalopathy.
Treatment
Successes in the understanding of pathogenesis led to attempts to use various drugs with immunosuppressive, anti-inflammatory, antifibrotic properties, as well as bile acids for therapy of patients PBC.
Glucocorticosteroids (GKS) appointed in a dose of 30 mg / day for 8 weeks. With a gradual decrease in the dose to 10 mg / day, lead to improvement clinical symptoms - temporary weakening of itching and / or increased fatigue, reduction of aminotransferase activity, IgG, but do not affect the level of serum bilirubin. GCS cause a decrease in the inflammatory response according to histology of the liver. With the continuation of placebo-controlled studies, for 2 years, there is no significant impact on the mortality rate. At the same time, after a year of therapy, a large problem was the potentiation of osteoporosis. Thus, GCS has the potential value for PBC therapy, however side effects Forcing them hazardous substances And do not appoint a long time at PBC. The risk of severe osteoporosis can be reduced during a combination of GKS with bifosphonates.
Budesonide represents the GCS of the second generation with low systemic activity, practically non-connecting side effects. The effectiveness of the drug in patients with PBC is carried out. There is reason to hope that this drug will be able to ensure all the advantages of the GCS, without exposing the life of patients with additional risk.
Cyclosporin A - major European tests involved 349 patients with subsequent observation of them up to 6 years (on average 2.5 years), did not confirm the prevention of histological progression of the disease or change the survival of patients receiving the drug. The high frequency of the occurrence of side effects, such as hypertension and deterioration of the renal function, does not allow the preparation for PBC therapy.
Azatioprin, chlorambucil, minority, D-penicillamine - due to the lack of a distinct effect on the progression of the disease and the presence of serious complications cannot be recommended for regular use in PBC.
Methotrexate at a dose of 15 mg orally 1 time per week can give a certain effect on clinical symptoms, bilirubinemia and SFF activity. However, in randomized controlled studies, the impact on the disease forecast was not detected. The pronounced side effects were noted.
Colchicine - a prerequisite for the use of the drug served as its antiofibrous and anti-inflammatory effect. The minimum toxicity of the drug led to the fact that the therapists recommend it for the treatment of PBC. In some cases, colchicine improves biochemical indicators. However, based on the results of randomized studies, it should be assumed that colchicine does not have any influence on cholestasis, histological progression or survival of patients.
The most promising drugs in the treatment of PBC should include ursodeoxycholic acid and adhemethionine.
Ursodeoxycholic acid (UDHK) is drug Treatmentwho has passed the most numerous studies of efficiency in the treatment of patients with PBC. Of all drugs pathogenetic therapy He is recognized most effective. Used at a dose of 10-15 mg / kg duration of 10 months. Up to 2 years and more, UPCC contributes to the oss of endogenous lipophobic toxic bile acids at the level of hepatocytes and biliary epithelium. Such a substitution of endogenous bile acids is due to competition between polar hydrophilic ursodeoxycholic acid and these acids during their transverseithelial transfer in the ileum. Reducing the number of potentially toxic endogenous bile acids against the background of cholestasis is accompanied by a decrease in cell membrane damage. In addition, UPCK is embedded in the phospholipid layer of the cell membrane, which leads to a direct stabilizing effect on hepatocytes.
The immunomodulating effect of the UPCM is carried out by reducing the expression of MNS IU and II classes on hepatocytes and epithelial cells of bile ducts, UDHK reduces the synthesis of IL-2, which leads to the suppression of stimulation citotoxic lymphocyte T-Helpers type 1.
Finally, the positive effect of UDHK is explained by its choleretic, hypocholesterolemic and litholithic effect.
UDCC contributes to a significant improvement in functional indicators, decreases or disappears skin itching. The effect on the morphological indicators is ambiguous, since in some cases they can progress.
A combined analysis of French, American and Canadian tests was carried out, which included 553 patients (276 received UDHK, and 277 - placebo). Average observation time - 4 years. The results of the analysis showed that on the background of UDCH therapy, liver transplantation was significantly longer. In multicenter trials in the United States, the observed survival after two years of the treatment of UDCH significantly exceeded the projected.
Resistance to the treatment of UPCH requires the exclusion of other causes of liver damage, and primarily cross-syndprom between PBC and autoimmune hepatitis.
In all studies conducted, it was noted that favorable effects are quickly achieved in the early stages of the cirrhosis; UDHK can be considered as a drug selection in treatment I-III stages PBC.
Ademethionine (S-adenosyl-L-methionine) is the initiator of three important methods of metabolism in the human body: polishing, rehabilfurication and polyamine synthesis. In these metabolic reactions, the drug is either as a donor of the methyl group or as an enzyme inductor.
One of the most important factors for regulating the functions of metabolism involved in the process of bile formation are the structure and composition of the hepatocyte membrane. With an intrahepatic cholestasis, a reduced viscosity of the membrane (a consequence of overpressure in it cholesterol) leads to a violation of the functioning of protein transport systems localized in it. Admethionine, participating in transmeteilication reactions, one of which is the synthesis of phosphatidylcholines, increases the mobility of the membrane and increases their polarization, which, in turn, leads to an improvement in the functioning of the transport systems of bile acids associated with hepatocyte membranes.
The liver transplantation is a method of choice for patients with progressive PBC and clinical and laboratory signs of hepatic decompensation. At the same time, a successful moment should be determined for operational intervention, since patients with terminal hepatic insufficiency "Big Surgery" is unacceptable. Disabled weakness, resistant skin itching, severe osteoporosis can be indicated for inclusion in the waiting list at earlier stages of PBC. Successful transplantation can completely return health for ten or more years, but sometimes the occurrence of PBC and in the transplanted liver is possible.
Literature
1. Ivashkin V.T., Buovers A.O. Autoimmune liver disease in the practice of clinician. Mr., Moscow 2001 102 p.
2. Pyymov S.D. Liver diseases. 3rd edition. Guide for doctors. M. Medicine .998. 703 p.
3. COOMBES B., CARITHERS R.L., MADDREY W.C. et.al. Hepatology 1995 v.22 p.759
4. Heathcote E.J., Lindor K.d., Poupon R., et.al. Gastroenterology 1995, V.108 A1083
5. Lombard M., Portmann B., Neuberger J., et.al. Gastroenterology 1993, v.104 p.519
6. Sherlock S., Dooley J. Diseasis of Liver and Billary System, 10th Blackwell Sci. Publication.-Oxford, 1997.-P.217-238
7. If the cause of the cholestase is not installed at the previous stages of diagnostic search and the AMA test negative test, the liver biopsy should be performed (III / C1).
8. In the case of a negative AMA of the test and liver biopsy data, comparable to PBC or PSX, is advisable, if possible, perform genetic analysis for research AVSV4 (The gene encoding the channel phospholipid export pump).
3. Primary biliary cirrhosis.
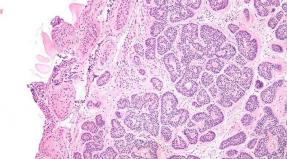
The disease can manifest weakness, itching and / or jaundice, but, as a rule, most patients are diagnosed on the asymptomatic stage. In rare cases, PBC is diagnosed at the stage of development of the complications of portal hypertension (ascites, hepatic encephalopathy, bleeding from varicosely extended veins of the esophagus). Usually, the diagnosis can be reliably installed with an increase in the level of SCF (hepatic origin) for 6 months and the availability of AMA in the diagnostic titrat. The diagnosis is confirmed by liver biopsy data with a pattern of non-aggated destructive cholangitis. At PBC, the level of the ICF andg. -GT. Transaminases and conjugated bilirubin may also increase, but these changes are not diagnostically significant. Typically improving the level of immunoglobulin M and cholesterol levels. In the detailed stages of the disease, a decrease in serum albumin levels, an increase in prothrombin time and bilirubin levels is noted. In 90% of patients with PBC, AMA is detected in the diagnostic titrat ≥ 1:40, their specificity is more than 95%. If possible, AMA-M2 is determined (antibodies to E2 subunit porvat dehydrogenase complex). In 30% of patients, PBC detect nonspecific antinuclear antibodies (Ana). Antibodies Anti - SP 100 and Anti - GP 210 have a specificity of more than 95% for PBC, these antibodies can be used as PBC markers in the absence of AMA. The sensitivity of these antibodies is lower than their specificity. Histologically allocate 4 PBC PBC stagesLudwig In accordance with the severity of damage to bile ducts, inflammation and fibrosis. Pathognomonic in the debut of the disease is considered a granulov detection in combination with the focal obliteration of bile ducts. The liver can be amazed unevenly, in one histological preparation there may be all 4 stages of the disease, for conclusion orient to the most pronounced changes. SPECIAL PBC ultrasound-signs do not exist.
1. To diagnose the PBC, it is necessary to increase the level of the SFC and the presence of AMA in the diagnostic titrat ≥1: 40 or AMA-M2. In this case, the conduct of biopsy liver is not mandatory, but allows you to estimate the activity and stage of the disease (III / A 1).
2. In the absence of specific antibodies, the liver biopsy is required to establish a diagnosis of PBC. With disproportional increase in the level of transaminase and / orIgG. biopsy is needed to identify concomitant or alternative processes (III / C 1).
3. Ama-positive patients with a normal level of liver tests should be observed annually with the study of biochemical cholestasis markers (III / C 2).
1. Patients with PBC, including patients with asymptomatic course of the disease, should receive ursodeoxycholic acid therapy (UDHK) at the rate of 13-15 mg / kg / day (I / A 1) Long (II -2 / B 1).
2. A good long effect of UDHK therapy is observed in patients in the early stages of PBC, as well as patients with a good biochemical response (II -2 / B 1), which should be assessed after 1 year therapy. The level of serum bilirubin ≤1 mg / dl (17 μmol / l) is considered a good biochemical response after 1 year of therapy, the level of ≤3 VGN and the AST level ≤2 VGN ("Paris Criteria") or a decrease of 40% or normalization of the level of the SFF ("Barcelona Criteria") (II -2 / B 1).
3. Currently, there is no consensus, how to treat patients with suboptimal biochemical response to UDHK therapy. It is proposed to use a combination of UDHK and Budesonid (6-9 mg / day) in patients on the docyrotic stages of the disease (stageI - III).
4. Liver transplantation should be considered in the terminal stage of the disease, when the level of bilirubin exceeds 6 mg / dl (103 μmol / L) or there is a decompensated liver cirrhosis with unacceptable quality of life or possible death during the year due to resistant ascitis and spontaneous bacterial peritonitis, recurrent bleeding Of the varicose extended veins of the esophagus, encephalopathy or hepatocellular carcinoma (II -2 / A1).
4. Driving PBC / AIG syndrome.
Primary biliary cirrhosis and autoimmune hepatitis are traditionally considered two different liver diseases. At the same time, there are patients with clinical, biochemical, serological and / or histological features of both diseases that can be detected simultaneously or sequentially. For these patients, the term cross syndrome is adopted. Etiology and pathogenesis of cross-syndrome are not quite clear. There is data on hereditary predisposition to autoimmune liver diseases. Each of the two diseases is induced by one or more trigger factors that launch the internal mechanisms of subsequent progression. When cross-syndrome, one or two unknown pathogenic factor is able to cause two different autoimmune liver diseases that flow at the same time. Either the only trigger factor is able to lead to a completely new immune response and as a result there may be a mixed picture of two autoimmune diseases with certain autoanthelters.
1. There are no standardized diagnostic criteria for cross-syndrome PBC / AIG. You should use the criteria shown in Table 4 (III / C 2).
2. You should always think about the cross-syndrome of PBC / AIG, when the diagnosis of PBC is set, because the therapeutic tactic will change when the diagnosis of cross-syndrome changes (III / C 2).
3. Combined therapy of UPC and Corticosteroids is recommended (III / C2). An alternative approach is the beginning of treatment with UPHK and in the absence of an adequate biochemical response within 3 months - the attachment of corticosteroids (III / C2). With a long-term course of immunosuppressive therapy, the dose of steroids can be reduced by adding Azatiotrian (III / C 2).
Table 4.
Diagnostic criteria for cross-syndrome AIG / PBC
______________________________________________________________________
Criteria PBC
1. SHF\u003e 2 VGN or γ GT\u003e 5 VGN
2. AMA≥ 1:40
3. Liver biopsy: Unnamed destructive cholangitis
Criteria Ayen
1. Alt\u003e 5 VGN
2. IgG\u003e 2 VGN or ASMA in diagnostic titra
3. Liver biopsy: Moderate or pronounced periseptal lymphocyte stepped necrosis
For the diagnosis of cross-syndrome AIG / PBC, at least 2 of the 3 criteria for each disease must be present. Mandatory the presence of typical histological data given in the AIG criteria.
5. Former sclerosing cholangitis.
Primary sclerosing cholangitis (PSX) is a chronic cholestatic liver disease, characterized by inflammation and fibrosis inside - and extrahepatic bile ducts. At PSX, the bile ducts are observed with the formation of multifocal strictures. PSX - progressive disease, ultimately leading to the development of cirrhosis and liver failure. The etiology of the disease is unknown, there is evidence of the participation of genetic factors in the development of PSX. The ratio of men to women among patients with PSX is 2: 1. As a rule, the disease is diagnosed at the age of about 40 years, although the diagnosis can be installed in childhood and old age. In 80% of patients with PSX there are inflammatory bowel diseases (BC), in most cases - ulcerative colitis. A typical PSX patient is a young man with a berth and / or clinical features of the cholestatic liver disease. In patients with clinical, biochemical and histological signs of PSX, but normal cholangiograms, the diagnosis of PSX small ducts is determined.
In half of patients, the disease is diagnosed on the asymptomatic stage. Typical signs: itching, pain in the right hypochondrium, weakness, weight loss, fever episodes. Less frequently, the disease manifests at the stage of cirrhosis and complications of portal hypertension. Physical examination most often reveals hepato - and splenomegaly. The most frequent biochemical feature of PSX is an increase in the level of the CFF. In the same time normal level SHF in the presence of a characteristic clinic should not exclude further diagnostic steps to establish the diagnosis of PSX. Often, transaminaz levels can be elevated 2-3 times from the VGN. In 70% of patients with diagnosis, serum bilirubin levels within normal values. 61% of patients have increased levelsIgG. , as a rule, 1.5 times from VGN. In patients with PSX, various antibodies are found: perinuclear antine-hydrophilic cytoplasmic (panca. ) (26-94%), antinuclear antibodies (Ana. ) (8-77%), anti-thunder antibodies (SMA ) (0-83%). To establish the diagnosis of PSP, the routine screening of antibodies is not required.
Liver biopsy data confirm the diagnosis of PSX, although they are not specific and differ in extreme variability. It is customary to allocate 4 PSX stages: portal, perportative, septal and cirrotic. For PSH, a specific picture of perideucatal concentric fibrosis is considered, but it can be detected far from always and cannot be considered pathogenic.
Ultrasound cannot serve as a choice for the diagnosis of PSX, but in some cases, experienced specialists can detect thickening and / or focal expansion of bile ducts. Typical cholangographic signs of PSX: diffuse multifocal ring-shaped strictures, alternating with areas of normal or slightly advanced ducts; short heavy-shaped strictures; Borrowed protrusion resembling diverticulus. As a rule, they are amazed inside - and extrahepatic bile ducts. At the same time, at PSS, an isolated lesion of intrahepatic bile ducts occurs (less than in 25% of cases). The Gold Standard of Diagnostics - ERCPG, however, this procedure may complicate the development of pancreatitis and sepsis. In some centers, the first step to establish the diagnosis of PSH is the MRHP. The sensitivity and specificity of MCHPG for the diagnosis of PSX is ≥80% and ≥87%, respectively. The MRHP better reveals changes in the ducts proximal to the obstruction site, and also allows you to detect pathology in the wall of bile ducts, assess the condition of the liver parenchyma and other organs. At the same time, small changes in the biliary tract in the debut of PSX can be missed with this study.
PSH in children. Diagnostic criteria are similar to those in adult patients with PSX. In 47% of cases, the level of the CFF may be within age norm, usually these patients have increased levels G. -GT. PSH debut in children is often characterized by the AIG clinic, including high levelsIgG, the presence of ANA and / or SMA In the diagnostic titrat and perportal hepatitis.
Differential diagnosis: PSX and secondary sclerosing cholangitis. To establish the diagnosis of PSX, first of all, it is necessary to eliminate the causes of the secondary sclerosing cholangitis: preceding operations on the biliary ways, cholangiolithiasis and the carcinoma of the bilia pathways, although it should be borne in mind that cholangiolithiasis and cholangiocarcinoma may complicate PSX currents. In the range of differential diagnostics should be includedIgG. 4-associated cholangitis / autoimmune pancreatitis, eosinophilic cholangitis, HIV-cholangiopathy, recurrent purulent cholangitis, ischemic cholangitis, etc. Differential diagnosis between primary and secondary cholangitis can be extremely difficult. Should take into account the features clinical flow Diseases, the presence of concomitant BCCs, changes detected on cholangiograms.
1. The diagnosis of PSX can be installed in patients with cholestase biochemical markers, typical MRHP data in the exclusion of the reasons for secondary sclerosing cholangitis (II -2 / B one). Conducting a liver biopsy to establish a diagnosis is optional, at the same time, biopsy data helps to assess the activity and stage of the disease.
2. With normal cholangiograms for the diagnosis of small ducts, the liver biopsy is required (III / C2). If there are significantly increased transaminases, liver biopsy data allow you to diagnose cross-syndrome AIG / PSH (III / C1).
3. ERCHP should be conducted (1) if the MRHP data is ambiguous (III / C2): The diagnosis of PSX can be set in the presence of typical changes to the ERCHP; (2) in a patient with a fever if there is a normal MRHP and suspicion of PSH (III / C2).
4. If the diagnosis of PSS is mounted to patients who have no instructions in the image, they should perform a biopsy colonoscopy (III / C1). In the presence of BCCs, patients with PSX colonoscopy must be repeated annually (in some cases - every 2 years) (III / C1).
5. To identify the formations of the gallbladder, an annual ultrasound conduct is required (III / C2).
6. Early diagnosis of cholangiocarcinoma Currently, according to the results of the study of biochemical markers or data of visualization methods is impossible. In the presence of clinical Indications ERCHP should be performed with brush cytology (and / or biopsy) (III / C2).
7. Reception UDHK (15-20 mg / day) improves liver samples and surrogate prognostic markers (I / B. 1), but does not have the proven influence on the survival rate of PSH patients (III / C 2).
8. Currently, there is no sufficient evidence base for the widespread use of UPCK as a chemopregnation of colorectal cancer at PSH (II. -2 / C2). Reception of UPCM can be recommended in high-risk groups: with hereditary exemplary for colorectal cancer, with preceding colorectal neoplasia or a long existing solved colitis (III / C2).
9. Corticosteroids and other immunosuppressants are shown only by patients with PSX / AIG cross syndrome (III / C2).
10. In the presence of pronounced strictures of bile ducts with significant cholestasis, the surgical expansion of bile ducts is shown (II -2 / B one). The installation of biliary stents and the drainage of bile ducts is performed with an unsatisfactory effect from the expansion of the ducts (III / C2). When conducting invasive interventions, preventive antibiotic therapy is recommended (III / C1).
11. In the terminal stages of PSX, the liver transplantation is recommended (II. -2 / A1), in the presence of dysplasia of cholangiocytes or recurrent bacterial cholangitis should also be given to the possibility of conducting liver transplantation (III / C2).
6. Cross-syndrome PSX / AIG.
This syndrome is an immuno-mediated disease and is characterized by histological features of AIG and typical changes for PSH changes on cholangiograms (III / C2). Forecast at the Cross Syndrome of PSX / AIG is better than with an isolated PSH, but worse than with AIG. It is recommended to conduct combination therapy of UPCC and immunosuppressants (III / C2). The terminal stages of the disease show liver transplantation (III / A1).
7. Immunoglobulin G. 4-associated cholangitis (F) .
Bilyiary liver cirrhosis is hard chronic illness biliary tract. The term "biliary" means that the destruction of the liver tissues is secondary in this case. First, the disease amazes bile ducts. This name unites two pathologies that develop for various reasons. Primary biliary cirrhosis (PBC) is an autoimmune disease, the cause of which the inadequate reaction becomes immune system On the fabric of the own body. Secondary cirrhosis has nothing to do with immune pathologies and develops from those patients who had previously experienced difficulties with bile outflow. Accordingly, the methods of etiotropic treatment in two cases will differ, and symptomatic therapy may be similar.
Primary biliary cirrhosis
Primary biliary cirrhosis of the liver is a chronic inflammatory-destructive pathology of autoimmune origin, in which intrahepatic bile ducts are destroyed. Due to the inability to exit bile, cholestasis and liver cirrhosis develops. Cholestasis is called stagnant bile and poisoning of the body with its products. Cirrhosis is a state at which normal functional hepatocytes are replaced by a dense connective tissue. In the development of this disease, the processes of necrosis (dieting) of tissues prevail.
Causes of development
PBC is a disease of autoimmune origin. The exact reasons for its development could not find out, but a genetic predisposition to primary cirrhosis was noted. In many cases, the members of one family are treated with similar symptoms. Also in the blood of patients were found antibodies that detect with autoimmune processes.
The essence of such processes is reduced to the fact that the human immune system reacts inadequate to its own organs and tissues. The main task is to allocate protection factors that are needed to be involved in the fight against alien agents. Such protective proteins are called immunoglobulins, or antibodies. They interact with antigens - proteins that distinguish pathogenic microbes and neutralize them. In case of autoimmune diseases Similar factors of protection are distinguished, but they are aimed at the destruction of non-infection, but their own cells and body tissues.
Primary biliary cirrhosis of the liver rarely develops as an independent disease. In disruption of the work of the immune system, the disease affects not only biliary pathways, but also other organs. At the same time, you can diagnose diabetes mellitus, glomerulonephritis, vasculitis, rheumatoid arthritis, red systemic lupus, dermatitis and other manifestations of immune reactions.
Symptoms
The symptoms of the primary biliary liver cirrhosis may not appear for a long time. The course of the disease can be asymptomatic, slow or progress quickly. At the stage, when clinical signs are not manifested, the only method of diagnosing the disease becomes laboratory methods. The most informative is the blood test with the definition of biochemical indicators and specific antibodies.
Characteristic features for which the primary biliary cirrhosis can be suspected:
- skin itching and rash;
- jaundice;
- general deterioration of well-being;
- digestion disorders;
- pain in the right hypochondrium.
The next symptom, which makes clarity into the mechanism of development of the disease, is jaundice. Syndrome is associated with the accumulation of bile and the inability to go to duodenal gut. Normally, bile is formed in the liver and flows on bulls in gall-bubble. This body serves as a kind of reservoir, and the bile is derived from them in the intestinal ducts only when the digestion process begins.
In the destruction of intrahepatic ducts, bile does not have the ability to get into the intestine. The problem is that it has a very aggressive environment for digesting food. If you get into the tissue of a person, it causes them necrosis. The most toxic bile product is bilirubin. He gives her a kind of amber color, and also stains calories and urine. If it gets into the bloodstream, the skin and mucous membranes of the patient acquire a yellow shade. This phenomenon is called jaundice. It often manifests itself no earlier than 6-18 months after the start of the skin itch.
With such violations, the digestive tract cannot but suffer. Patients complain of dyspepsia, pain in the stomach and intestines. The main symptom, which indicates the destruction of the liver, is an acute pain in the right hypochondrium. Instrumental research (ultrasound) will help determine that the liver is inflamed and increased in size. The spleen in most cases remains unchanged.
Stages of primary cirrhosis
Diagnose the stage of primary biliary liver cirrhosis can be only based on biopsy data. Other methods, including blood test, will not be able to distinguish the intrahepted stagnation of bile from the extraordinary one. It is important that in autoimmune cirrhosis, it is an intra-brandy cholestasis.
- The first stage is portal, there is inflammation and destruction of the structure of interdolkovoy and septal bile ducts. The surrounding liver tissues are also inflamed and impregnated with infiltrate with leukocytes. Stagnation of bile at this stage is not possible yet.
- The second stage is periportal, primarily characterized by further inflammatory changes in bile ducts and hepatic tissue. The number of functioning ducts decreases, and therefore the outflow of bile is already difficult. You can detect signs of cholestasis.
- The third stage is septal. Tight connective tissue scars are formed between portal paths, bile strokes and hepatic veins. Changes affect a healthy liver parenchyma, as a result of which it is replaced on a scar cloth. Around these heavys continues inflammatory process With lymphocyte infiltration.
- The last stage is actually cirrhosis. In the liver, small nodes (micronodule cirrhosis) are formed. A normal liver structure is disturbed, as a result of which it is not able to perform its functions. Signs of central and peripheral stagnation of bile are found.
Depending on the stage of primary cirrhosis, you can determine the forecast. The earlier it will be possible to put the correct diagnosis and begin treatment, the greater the patient has a chance of recovery. The clinical picture of the cirrhosis is already incurable pathology, in which it is about maintaining the state of the patient and an increase in life expectancy.
Diagnostic methods
The most affordable method of diagnosis is a blood test. In suspected primary biliary cirrhosis, biochemical analysis is carried out, as well as immunological tests for detecting antibodies. The overall picture will look like this:
- raising the level of free bilirubin;
- reducing the number of common protein;
- increasing the concentration of immunoglobulin A, M and G;
- identification of mitochondrial antibodies (AMA), as well as antinuclear (ana);
- increased copper content and reduced - zinc.
Ultrasound and MRI liver in this case are non-informative. They will not be able to identify the pathology of intrahepatic bile ducts. Changes in primary cirrhosis should be distinguished from similar diseases that proceed with bile stagnation syndrome: viral hepatitis, stricture of biliary tract, liver neoplasms, intrahepatic biliary tumors.
Treatment and forecast scheme
Effective treatment of primary biliary liver cirrhosis is currently not developed. All methods are reduced to the elimination of symptoms and improving the quality of the patient's life. They include drug therapy and diet, common to all diseases with bile stagnation.
Immunosuppressors, anti-inflammatory and antifibrotic agents, as well as bile acids are prescribed from the drugs. To remove the skin, use sedatives and UFOs, and with a deficiency of vitamin D - vitamin complexes With its content. At the last stages, medication therapy will not be effective. The only opportunity to extend the life of the patient and continue to treat it - this is a liver transplant from a healthy donor.
The forecast depends on the stage of the disease, the timeliness of the treatment and the associated symptoms. With a latent (asymptomatic) patient's course, it can live 15-20 years old and more if you follow the recommendations of the doctor and take drugs. If clinical signs have already manifested themselves, life is shortened to 5-10 years. With liver transplantation there is a risk of recurrence. This occurs in 15-30% of cases.
Secondary biliary cirrhosis
Secondary biliary cirrhosis of the liver is a chronic progressive disease of the liver, which develops in pathologies of extrahepatic biliary tract. The final stages are also characterized by fibrous restructuring of the liver parenchyma, as a result of which it loses the ability to perform their work. With this disease it is necessary surgeryTo eliminate the mechanical obstruction of the biliary tract.
Causes of development
Secondary biliary cirrhosis develops when blocking (obstruction) of the main bile duct or its large branches. Thus, the bile cannot flow into the intestines, poisoning with toxins the body and causes inflammation of the surrounding tissues.
Pathologies that can lead to biliary cirrhosis:
- cholelithiasis;
- consequences of the operation to remove the gallbladder;
- in rare cases - benign tumors and cysts;
- in children - congenital atresia of the biliary tract, fibrousosis.
Since bile does not have exit from bile ducts, their walls in some areas are expanding. Pathological tanks for bile are formed, around which inflammatory processes begin. The functional parenchyma liver is damaged, and instead of normal hepatocytes, connective tissue islands are formed. This process characterizes the development of micronodule (fine-income) cirrhosis.
Symptoms
Symptoms of biliary liver cirrhosis are similar to the primary type. You can distinguish these two diseases according to the results of blood tests and a biopsytte study. Most patients appeals to a doctor with similar symptoms:
- pronounced skin itching;
- jaundice;
- pain in the right hypochondrium;
- fever, nausea, general worsening of well-being;
- reduced body weight;
- pottening urine and light colors.
The mechanisms for the development of characteristic clinical signs are similar. At the later stages, the disease progresses, dangerous symptoms of hepatic insufficiency begin to manifest. The danger to the life of the patient is abscesses that are formed in the liver parenchyma.
Methods of diagnosis and treatment
In the primary inspection, the doctor is important to know whether the patient's patient has a disease of biliary tract or an operation to remove the gallbladder. At palpation, the increase in liver and spleen, soreness in the field of right hypochondrium is noted.
No less informative will be ultrasound diagnostics. In some cases, it is possible to detect the cause of the blockage of the biliary tract (stones, tumor) and the exact localization of an outsider. The final diagnosis is made on the basis of percutaneous cholangiography - the studies of bile moves with the determination of their location of their blockage.
If the reason for the secondary biliary cirrhosis is revealed, there is no need for immunological tests. Nevertheless, the biochemical blood test will allow to obtain a more complete picture of the disease. Patients reveal the level of bilirubin, cholesterol, alkaline phosphatase, bile acids, decrease in the amount of protein. The overall blood test will show increase in leukocytes, anemia, an increase in the rate of sedimentation of red blood cells.
Treatment of biliary liver cirrhosis implies operation. If you do not remove an outsider, which makes it difficult for bile outflow, the disease will be progressing and further. There are various methods for performing such operations:
- choledochotomy;
- choledochostomy;
- removal of consolidation (stones) of their clouded ducts;
- svaldoch stenting under the control of the endoscope;
- dilatation (expansion) of bile ducts;
- their outdoor drainage.
The technique is selected individually. The surgeon must familiarize himself with the results of the tests, determine the localization of the pathological process and choose the most convenient access. Postoperative period Includes receiving medicines and diet. Just as with primary biliary cirrhosis, it is necessary to follow their health throughout life.
Before you figure out what biliary cirrhosis of the liver is, it is necessary to understand that this term combines two similar diseases with different etiology. They are both connected with the defeat of the bile ducts, the development of cholestasis and jaundice, as well as the destruction of tissues and the liver and the formation of scars in its parenchyma. Nevertheless, the causes of primary cirrhosis differ from those on which the secondary form develops. If the first option is associated with the lesion of intrahepatic ducts, then the second affects extrahepatic moves. Accordingly, the methods of diagnosis and treatment will differ, but they are reduced to one to elongate the symptoms of the disease and the elongation of the patient's life.
Total duration: 21:51
Alexander Sergeevich Trukhmanov, Doctor of Medical Sciences, Professor:
Let me with pleasure to give the floor to the doctor of Medical Sciences of Shirokova Elena Nikolaevna with a message "Modern consensus on the diagnosis and treatment of primary biliary cirrhosis and primary sclerizing cholangitis." Please Elena.
Elena Nikolaevna Wisdom, Doctor of Medical Sciences, Associate Professor:
Many thanks, Alexander Sergeevich.
Let me introduce you modern condition The question of the diagnosis and treatment of primary biliary cirrhosis and primary sclerosing cholangitis.
First of all, we will define that the primary biliary cirrhosis is. This is a chronic cholestatic liver disease, which is based on an imune-indirect destruction of small intrahepatic bile ducts. A characteristic feature is the presence of antimitochondrial antibodies.
The incidence of primary biliary cirrhosis ranges from 15 to 400 cases per million people. The overwhelming number of patients with primary biliary cirrhosis - about 90% are women. The average age of the manifestation of the disease is 50 years.
Currently, almost half of patients are diagnosed in the asymptomatic stage. In the absence of adequate treatment after 10 to 20 years, patients can develop cirrhosis of the liver and hepatic insufficiency.
A characteristic feature of the primary biliary cirrhosis is skin itching. Even more often than skin itching in patients with weakness. Moreover, there is no correlation of weakness with the severity of histological manifestations, with the severity of biochemical indicators of activity and with the age of the patient.
Half patients may have jaundice. Characterized by the presence of concomitant autoimmune diseases, such as the autoimmune damage of the thyroid gland, autimmune thyroiditis, Reino syndrome.
In some cases, we meet with pronounced skin hyperpigmentation, the presence of xantellasm and xanthma.
In 60% of patients, as a rule, increased in the size of the liver. According to biochemical samples, cholestasis is determined. The presence of antimitochondrial antibodies in the titer 1:40 and more is characteristic sign.
What the morphological data is swinging, then the presence of non-aggregative destructive cholangitis is determining.
In this slide, you see a photo of our patient, which suffers primary biliary cirrhosis. Pronounced xantellasm and xanthoms that are less common. Approximately 10 patients with severe cholestasis, they are located on the back surface of the brushes and at the level of the elbow bend. This is due to an increase in serum cholesterol over 400 mg / dl, if it is observed for more than three months.
So, what are the main diagnostic criteria of primary biliary cirrhosis. It is an increase in the level of alkali phosphatase (SCF) and gammaglutamyltranspendase, the presence of antimitochondrial antibodies of the M2 fraction, aimed at the E2 component of the piruvat-dehydrogenase complex. This is the presence of destructive cholangitis, lymphocytic infiltration.
In some cases, approximately 10 - 20 patients who suffer from primary biliary cirrhosis, we faced the situation when there are features and autoimmune hepatitis. We are talking about the so-called phenomenon of the cross. Crossback syndrome is a combination of signs and autoimmune hepatitis and primary biliary cirrhosis.
It is believed that for the formulation of this diagnosis, it is necessary to have two of the three criteria listed here for each disease.
For primary biliary cirrhosis it is:
- increased alkali phosphatase level by more than 2 times from the upper limit of the norm, or the level of gamma-glutamylTranspend of gamma more than 5 times from the upper limit of the norm;
- the presence of antimitochondrial antibodies in the titer 1:40 and above;
- the presence of non-native destructive cholangitis according to the liver biopsy.
For autoimmune hepatitis, the presence of the following criteria:
- an increase in the level of alanine transaminase is more than 5 times from the upper limit of the norm;
- improving the level of immunoglobulin class G more than 2 times or the presence of antibodies in smooth muscles in the diagnostic titer 1:80;
- according to the liver biopsy, it is important to determine the perioportal or periseptal step necrosis.
Histological preparation. This is the liver tissue of our patient, which suffers from the crossing syndrome (primary biliary cirrhosis and autoimmune hepatitis). Here expressed lymphogystocyte infiltration in the portal tract, in the center of the presence of stepped necrosis. Slightly rightly unevenly expanded lumen of the yolk duct (the phenomenon of the proliferation of the doors).
It is well known that the drug that is officially approved in all countries for the treatment of primary biliary cirrhosis is "ursodoxicole acid" (UDHK). Interesting data Pares A., which were presented in the journal "Gastroenterology" in 2006, where the influence of "ursodeoxycholic acid" was assessed on the survival of patients with primary biliary cirrhosis.
The survival rate of those patients who had a good response to therapy was actually no difference from the age and population. Specificably exceeded the survival rate, which was projected by the Mao model. This is a green "curve." These data is reliable, and the survival of patients with a good biochemical response is straightely different from survival, which is predicted according to the Mao model. And the Mao model is a practically basic model that allows you to calculate the prognostic survival rate of patients with primary biliary cirrhosis.
What to consider a good biochemical answer. It was accepted to determine after the year of the therapy "Ursodezoxycholic acid". There are so-called Paris criteria. This means - normalization of the level of bilirubin. It should be less than 1 mg / dl (or less than 17 μmol / l) in the SI system.
The level of alkali phosphatase (SCF) must be less or equal to a three-time limit of the norm. The level of aspartaminotransferase (AST) must be less than or two norms.
As for Barcelona criteria, this is a decrease of 40% or normalization of the level of alkaline phosphatase in a year of the therapy "Ursodezoxycholic acid".
We have your own experience of four-year-old "Ursodoxicole acid" therapy, Ursosan drug in patients with primary biliary cirrhosis. We have shown that on the effect on the biochemical parameters of Ursosan is most effective in patients with the first stage of primary biliary cirrhosis. It was for them that the normalization of the level of serum transaminases and a decrease in the level of bilirubin by more than 2.5 times. Bilirubin is the main prognostic marker in patients with primary biliary cirrhosis.
The minimum therapeutic effect was noted in patients with the fourth (last) stage of the disease at the liver cirrhosis stage, which is consistent with the data of international studies.
So, the strategy is such. Patients with primary biliary cirrhosis should receive "ursodoxicole acid" in a dose of 13 - 15 mg / kg / day. This is standard, officially approved therapy.
If the biochemical response is observed, which we have already spoken earlier, it is necessary to continue the monotherapy "Ursodezoxycholic acid" under the constant control of the state of the patient, the level of biochemical samples.
If there is no answer, and there are signs of crossover with autoimmune hepatitis, the phenomenon of lobular hepatitis, an increase in the level of aspartic transaminase, or the other situation, then a suboptimal biochemical response is obtained. Complete answer we expected, we do not get. This is almost a third of patients.
What to do. In this situation, the only universal strategic step is currently not developed. Various options are assumed. One of them is an additional appointment "Budesonide" at a dose of 3 to 9 mg per day.
The preparation of the second stage is "Mikophenolate Mofetil". This is an immunosuppressive therapy that allows you to level or reduce side effects from corticosteroids. The proposed dose of half a gram per day.
If there is no answer, now the question of the possibility of applying fibrats is being considered. The duration of this course is currently not yet defined. The estimated dose of 200 mg of the drug per day.
So, what recommendations for the treatment of primary biliary cirrhosis can be formulated today. According to European society According to the study of liver disease, it is believed that the formal approved is "ursodeoxycholic acid". Dose 13 - 15 mg / kg / day for a long time. With suboptimal biochemical response, a combination of "ursodeoxycholic acid" can be combined with "budesonide" (second-generation glucocorticoid).
As for cross-syndrome, here it is possible, and a combination of "ursodoxicole acid" with corticosteidia is required. With the second embodiment, the monotherapy "Ursodoxicole acid".
In our clinic, which is headed by Academician Vladimir Trofimovich Ivashkin, we have our own good experience of treating patients with crosstrozoxycholic acid cross-syndrome with corticosteidia.
Our patients (58 patients) were divided into 2 groups in accordance with the cross-syndrome option. Patients with the first option were taken by corticosteroids and Ursosan (URSO-oxidic acid - in a standard dose of 13 - 15 mg / kg / day).
The second option is patients who have histological features resembling primary biliary cirrhosis. At the same time, they had antibodies in smooth muscles and antinoclorenic antibodies in the diagnostic titrat and highly high biochemical activity, raising the level of transaminase. They received the Ursosan monotherapy.
In 60% of our patients, a full response was observed, and more than a quarter showed a partial response to therapy.
When analyzing the cumulative survival of patients with cross-syndrome, we obtained that the survival rate of patients exceeds the survival rate, which was predicted according to the Mao model. The survival of our patients is the top yellow "curve." The lower red lens is the survival rate that is projected on the Mao model. Ursodeoxycholic acid can improve the survival rate of patients with cross-syndrome.
What new directions in the treatment of primary biliary cirrhosis currently exist. These are agonists of a farneseoid X-receptor (FXR) - "obtashephole acid". Is it 6? ethyl-minesoxycholic acid, which is now going through the third stage clinical studies. It can be previously said that it improves the biochemical tests of patients with primary biliary cirrhosis and reduces the level of serum immunoglobulin M.
And the second direction is PPAR agonists?. These are fibrats. They have anti-inflammatory and immunomodulatory property. Currently actively studied.
The second direction of my today's message is the primary sclerosing cholangitis. It is also a chronic cholestatic liver disease, which is characterized by diffuse inflammation and fibrosis of intra and extrahepatic bile ducts.
Unlike primary biliary cirrhosis, primary sclerosing cholangitis is predominantly male. The ratio of men to women 2: 1. As a rule, the disease is diagnosed in patients aged 40 years. Extremely rarely - in children. In 60 - 80,% of cases there is a combination of primary sclerosing cholangitis with inflammatory bowel diseases. 80% are patients with nonspecific ulcerative colitis, 10 - 15% is the Crohn disease.
Various are possible clinical options Debut primary sclerosing cholangitis. It may be an asymptomatic increase in the level of hepatic samples. The patient passes a survey in the framework of the closerization and it records elevated cholestasis syndrome markers.
Either this is a classic manifestation (skin itching, weakness, jaundice). Or it may be markers of recurrent bacterial cholangitis. Either the diagnosis passes already at the stage of complication of cholestasis. Either at the stage of complication of portal hypertension, when the debut occurs with bleeding from the varicose extended veins of the esophagus.
Most often we record an increase in alkaline phosphatase. As a rule, it is a 100% discovery in a biochemical blood study. Asparagic and alanine transaminases are elevated almost in 90% of patients. Gamma-glutamilTransferase in 85% of cases.
Antinerophilic cytoplasmic antibodies (ANCA) are detected in 65 - 70% of cases (especially if the patient has a non-specific ulcerative colitis). In 60%, bilirubin can be increased. Antibodies in smooth muscles, antinuclear factor we encounter approximately half of patients.
The main diagnostic criteria of primary sclerosing cholangitis. This is the presence of chronic cholestasis, that is, an increase in the level of gamma-glutamytranspendase, alkaline phosphatase, lucinopeptidases (laps). This is the data of endoscopic retrograde cholangiopancratography or magnetic resonance cholangiography. Of course, the elimination of the causes of the secondary sclerosing cholangitis.
Changes that are typical when conducting cholangiography. This is the presence of diffuse multifakal ring-shaped strictures, which alternate with the plots of normal or slightly defined ducts. The presence of short tight strictures or fabriced protrusions that resemble diverticulus.
These endoscopic retrograde cholangiopancratography. Arrogments marked strictures of extrahepatic bile ducts.
Magnetically resonant cholangiogram of the patient of 72 years, which suffers primary sclerosing cholangitis. The upper arrow demonstrates a narrowing at the level of the front right hepatic duct, and the lower arrow indicates the place where the general liver duct should be visible. Lack of visualization speaks about the presence of stricture.
As for the liver biopsy data, there is a typical sign here - "Lukovichnaya husk". This is the presence of concentric fibrosis. But when the question arises, whether patients need to perform a liver biopsy, currently such recommendations are: no, not to all patients.
If you have no doubt a diagnosis of primary sclerosing cholangitis, there are typical biochemical features, typical cholangiogram data, then in this case, morphological verification can wait.
If you have suspicions that there is a cross-syndrome in conjunction with autoimmune hepatitis, or you suspect a sclerosing cholangitis in small ducts (when there are no characteristic cholangographic data), then it is definitely a decisive word for the liver biopsy.
Ursodeoxycholic acid is one of those drugs that intensively, actively and widely studied in the therapy of patients with primary sclerosing cholangitis. It is well known and approved for the treatment of primary biliary cirrhosis. Given the similarity clinical manifestationsMany researchers have tried this preparation in the therapy of primary sclerosing cholangitis.
What features, what actions of the drug can be considered attractive. "Ursodeoxycholic acid" stimulates the detoxification processes of bile acids, stimulates secretion, has an overwhelming apoptos property. In addition, protects the cholangiocytes from toxic action hydrophobic bile acids. Even the antifibrotic effect of the drug is described.
Primary sclerosing cholangitis. Lindor research data in 1997. 105 patients entered the study. "Ursodezoxycholic acid" was used in a standard dose of 13 - 15 mg / kg for 2-5 years. An improvement in biochemical parameters in patients with primary sclerosing cholangitis was noted. At the same time, there was no reliable influence on clinical signs, noted.
Olsson data, 2006th year. More representative patient cohort, higher dose of the drug. "Ursodeoxycholic acid" was taken at a dose of 17-23 mg / kg / day for five years. There was an excellent tendency to improve survival against the background of the "Ursodoxichetic acid" admission. However, it was not statistically reliable.
According to the Pilot Research, Mitchell was noted good tolerability of the drug at a dose of 20 mg / kg / day. Improving hepatic samples was noted. A large representative study was held in the USA, which was attended by 150 patients. There was a higher dose of the drug (28 - 30 mg / kg / day). For five years, patients had to take this drug.
However, the study was discontinued early, because in a group that received "ursodoxicole acid", more private fatal outcomes were observed, the need for liver transplantation or fatal outcome.
There are interesting data that Ursodeoxycholic acid is able to reduce the risk of developing colorectal dysplasia in patients with primary sclerosing cholangitis and non-specific ulcerative colitis. The experiment showed that "deoxycolic acid" stimulates the proliferation of colorectal epithelium in animals. In turn, "Ursodoxicolithic acid" suppresses apoptosis, which is induced by "deoxycolic acid". "Ursodezoxycholic acid" inhibits the growth of intestinal cancer cells, which are stimulated by "deoxycolic acid".
At the same time, the basis for the general recommendations of the unconditional admission of "ursodeoxychole acid" in patients suffering from primary sclerosing cholangitis currently not. As for the recommendations of the European Society for the Study of Liver Diseases, it is proven that the reception of the drug in a dose of 15-20 mg / kg / day improves liver tests and prognostic disease markers. However, the effect on survival was not proved. For the prevention of colorectal cancer, the drug can be recommended in high-risk groups.
Bilyary cirrhosis is a liver pathology, which develops against the background of the difficult outflow of the bile both inside the liver and in the extra-artistic biliary tract. The greatest number of patients with this disease - adults after 25-30 years, in childhood The disease is extremely rare.
If we consider overall statistics of the cirrhosis, the biliary lesion of the liver is diagnosed in about 10 cases out of 100. Bilyary cirrhosis is considered the most poorly studied, therefore it is necessary to consider the features of its development and treatment for each of the forms of pathology.
Bilyary cirrhosis is a very rare form of pathology, so it is not always possible to quickly make the correct diagnosis. In most cases, over a long time, the disease proceeds asymptomatic and is found randomly during the dispensarization or during the diagnosis of other diseases. The symptoms of biliary cirrhosis usually occur when the disease goes into a heavy stage, and in addition to the body transplantation, the patient cannot be helped by anything.
Bilyary cirrhosis is characterized by substitution of healthy fibrous tissue. This happens when the affected parenchym cells are not able to cope with their functions.
The more liver cells are affected, the more pronounced liver failure and the higher the probability of complications of complications: portal hypertension, ascites and lesions of other internal organs.
Life expectancy with such a diagnosis directly depends on the stage on which the disease was detected. Cases are registered when patients for two decades have not been suspected of the pathological lesion of the liver, and also known the rapid development of the disease, when the fatal outcome stepped within 2-3 years after the start of the development of the cirrhosis.
Moreover, the rate of development of the disease and the growth of fibrous tissue in each patient differs and depends on the set of factors: the state of the immune system, the age of the patient, its lifestyle and the presence of concomitant diseases. It is possible to predict the development of the disease only after a complete examination of the patient, taking into account various factors.
Bilyary cirrhosis is made to divide into two forms - primary and secondary, each of which has its own characteristics. The development of primary form is indicated when the disease develops under the influence of autoimmune factors and initially leads to the development of cholestasis and only then goes into cirrhosis of the liver.
Secondary biliary cirrhosis of the liver is the consequence of chronic inflammatory processes associated with violation of bile outflow. But independently of the form and causes of the disease, biliary cirrhosis has both general features and symptoms.
Primary form of the disease
So far, despite many studies, it was not possible to identify the exact causes of the development of the primary form of biliary cirrhosis. It is only known that the damage to the liver cells occurs under the influence of T-lymphocytes, the functions of which are aimed at suppressing the livelihood of foreign particles in the body. But for any reason, T-lymphocytes begin to count the cells of the body are dangerous and begin to destroy them.

T-lymphocytes initially begin to affect small bile ducts, leading to their destruction and development of cholestasis. Due to the delay in bile, liver cells begin to suffer from toxic lesion, as a result of which the inflammatory process begins in the liver. The affected hepatocytes are replaced by a fibrous cloth, which forms scars in the organ. It is noticed that the larger the fibrosis of the liver progresses, the less pronounced the inflammatory process becomes.
Stages
It is customary to allocate 4 stages of development of primary biliary pathology:
- The first - there is inflammation of interdollak and septal channels, which is accompanied by the extension of the vessels. There is lymphocytic infiltration to form a granul.
- The second - the inflammatory process goes to the liver parenchyma, going beyond the boundaries of the portal paths. The defeat of most ducts is observed, and the remaining bile ducts have an abnormal structure.
- The third - progressive inflammation leads to a more pronounced cholestase, and spikes from connective tissue are formed in the parenchyma.
- The fourth is characterized by the lack of ducts in portal aisles, the process of liver cell necrosis begins.
The reasons that lead to a failure in the work of the immune system are unknown. But many scientists are inclined that there is a conflict between lymphocytes and histocompatibility antigens, characteristic of the "transplant against host antigens", since the zirrosis development mechanism is very similar to the processes occurring at such a reaction, but this version is still under consideration.
Like any autoimmune disease, biliary cirrhosis in 90% of cases amazes women after 30-40 years. That is why there are versions that the reasons are hormonal restructuring in the body, as well as the physiological wear of the body. The biliary cirrhosis of the primary form has properties to spread in a circle of one family, which confirms the hereditary predisposition to the disease.
Alla writes: "Mom was diagnosed with" primary biliary cirrhosis ". The doctor suggests that the cause is blood transfusion. It is after this procedure that problems began with thyroid gland and joints. "
Symptoms
Along with the biliary lesion of the liver, the concomitant development of other diseases of autoimmune origin is characteristic:
- System red lupus.
- Sclerodermia.
- Rheumatoid arthritis.
- Vasculitis.
- Glomerulonephritis.
- Schogen syndrome.
- Autoimmune thyroiditis.
At the very beginning of the development of the disease, symptoms appear only in a small number of patients. In most patients, clinical signs occur only with the extensive growth of fibrous tissue.

The very first and characteristic feature is the skin itching, resulting from a large number of bile acids, which annoyingly acting on the nerve endings. Sometimes itching is initially accompanied by a jaundice, but it may occur at later stages. Experts argue that the later the yellowness of the skin appears, the more favorable disease forecast.
Vascular stars and "liver palms" with this form of the disease are extremely rare. Half patients appear hyperpigmented stains in the joints of the joints of the joints, and after - and other parts of the body. At the later stages, pigmented fate of the skin thicken, and the external clinical picture resembles focal sclerodermia.
For biliary cirrhosis, xantellasm in the field of eyelids, chest, elbows and knee joints is characteristic.
Other symptoms:
- The increase in liver and spleen in size is occupied about 60% of patients.
- Dyspepsic disorders, bitterness in the mouth, pain in the right hypochondrium.
- General weakness, lack of appetite.
- Dry skin.
- Muscular and articular pains.
- Subfebrile temperature increase.
When progressing cirrhosis itch becomes constant and unbearable. Evenum appears, ascites develop, and in the esophagus, internal bleeding can occur in the esophagus.
Diagnosis and treatment
Diagnosis of biliary cirrhosis is based on data biochemical analysis blood, identification of antimyolochondrial antibodies and instrumental methods - ultrasound, CT and MRI liver. In the primary biliary cirrhosis, the activity of hepatic enzymes increases, ESP and the concentration of bile acids increases. Antimochondrial antibodies are found in almost every patient, and approximately half of the halves there is an appearance of a rheumatoid factor and antinuclear tel.
The liver biopsy is necessary to confirm the diagnosis, early detection of destructive cholangitis and identify the specifics of the development of cirrhosis in heavy stages.
Primary biliary cirrhosis is dangerous in that there are no special preparations for its treatment, so all therapeutic measures are directed to the removal of symptoms. First of all, patients prescribe a strict diet:
- No more than 40 g fat per day.
- Use of protein 80-120 g per day.
- Failure to food containing preservatives and dyes.
- Exclusion of alcoholic and carbonated drinks, strong tea and coffee.
- Doctors recommend to adhere to the diet number 5 and drinking regime - 1.5-2 liters of clean water per day.

Diet "Table No. 5"
What drugs are prescribed:
- Cyticostatics (hexal).
- Corticosteroids (prednisone).
- Bisphosphonates (alendronat).
- Hepatoprotectors (essential, phosphoglie, hepaben).
- Choleretic (Allohol).
The means of overwhelming the synthesis of collagen - Cupinenil, Dr. Penicilline can be selected. Ursosan, rifampicin and phenobarbital are suitable for removing itching. The only method with which you can cure a disease - a transplant of the donor body.
Opinion of a specialist: "Liver transplantation is effective only at the compensated stage. When decompensation, such operations are not carried out, since they are meaningless. "
Secondary cirrhosis
Secondary biliary cirrhosis, unlike primary, more studied and understandable. It develops, with chronic joy of bile in the paths located inside and outside the liver. What leads to secondary biliary cirrhosis:
- Congenital deviations in the development of biliary tract.
- Cholecystolithiasis.
- Cholestasis.
- Cysts and other benign neoplasms.
- Cancer tumors in the pancreas.
- Squeezing the bilny channels with increased lymph nodes (lympholecosis, lymphogranulomatosis).
- Purulent or primary cholangitis.
- The narrowing of the biliary channels after surgical intervention.
- Cholelithiasis.
These pathologies lead to a long-term stagnation of bile and increase the pressure in biliary ducts, which they begin to swell. Chronic flow Diseases provokes the exhaustion of the walls of the ducts, and the bile penetrates the liver parenchyma. Under the influence of acidic and aggressive fluid, the liver cell is inflamed, and the necrosis process begins.
The affected hepatocytes are gradually replaced by fibrous fabric. The speed of this process is different - on average from 6 months to 5 years. The process is accelerated if bacterial infection is joined or complications are developed. The disease leads to a rack of hepatic insufficiency, against the background of which the last stage is developing - the hepatic coma.
Manifestations
Symptoms of primary and secondary biliary cirrhosis have a lot in common. But the secondary liver damage occurs with an equal frequency in both sexes, while the primary form is more characteristic of female.
Clinical signs of disease progression:

In recent stages, signs are joined:
- portal hypertension;
- ascites;
- varicose veins of the veins of the esophagus and intestines.
Diagnosis and therapy
The diagnosis of secondary biliary cirrhosis is to collect anamnesis, patient complaints and its inspection. After that, the following surveys are prescribed:
- Blood and urine tests.
- Uzi liver.
- MRI and CT.
For the disease, promotion is characterized by:
- blood sugar;
- alkaline phosphatase;
- cholesterol;
- bilirubin; Alt.
Most patients are diagnosed with eosinophilia, anemia and an increase in ESP. Be sure to assess the amount of copper in the urine - high content speaks from the severity of the process. In definitely, it is necessary to diagnose the detection of HCB, cholecystitis, cholangitis, pancreatic damage. But the most accurate diagnosis is made by taking a biopsytte and histological research of the material.

It is possible to delay the progression of the disease, if we exclude the causes of stagnation of bile. Therefore, it is very often resorted to surgical interference to remove stones or stenting a duct. The liver transplant does not always give a positive result, the patients have a reuse of the disease.
If it is impossible to carry out surgery, hepatoprotectors, vitamins, antioxidants, antihistamines and antibiotics are prescribed to prevent the development of bacterial infection.
Eugene writes: "After removing the gallbladder, the stomach was constantly sick, there was bad well-being. But the doctor reassured that it was simply "postcholecistomic syndrome," you need to maintain a diet and everything will pass.
After a few months, he went to another doctor, where they found the narrowing of the biliary tract from abroad and strong inflammation. The doctor said that if he had come earlier, it would be possible to stop the process, and now I have a precross a state that is rapidly progressing. "
Disease development in children
Cirrhosis in childhood is not uncommon, but the biliary form is practically not found in childhood. Primary biliary cirrhosis is usually developing in middle-aged patients, but the secondary form of the disease may arise due to the abnormal development of biliary tract and in children.
The main causes of children's biliary cirrhosis are fibrousosis and artisia of the biliary tract. As in adult patients, the disease is developing due to the disturbed outflow of bile, after which the cholestasis is developing with the transition to the cholangitis, which leads to the liver cirrhosis.
Treatment of biliary cirrhosis in childhood requires the intervention of experienced specialists and continuous support of the diet. With the unfavorable development of the disease, a liver transplantation is carried out.
Forecasts and complications
Primary biliary cirrhosis is primarily dangerous in that it is impossible to establish the cause of the disease, therefore there are no specific treatment methods. Doctors recommend eliminating all the factors that can affect autoimmune processes:
- Exclude physical and nervous overvoltage.
- Avoid stressful situations.
- Heat the foci of infection.
- Normalize hormonal background.
Primary and secondary biliary cirrhosis have general complications:

Primary biliary cirrhosis is often complicated by concomitant autoimmune diseases: systemic lupus, scleroderma, rheumatoid arthritis and others.
Skin covers are very often affected by primary form, except for jaundice and hyperpigmentation, vitiligo is often observed - the appearance of white unplanned skin sections.
Life expectancy depends on a variety of factors, but based on statistical data, you can define common indicators:
- Primary shape with bilirubin level up to 100 mkmoll / l - about 4 years of life, over 102 μmol / l - no more than 2 years.
- Revealed in the early stages and uncomplicated primary cirrhosis - about 20 years.
- Secondary biliary cirrhosis with pronounced symptoms - 7-8 years.
- The asymptomatic flow of secondary cirrhosis increases the lifespan until 15-20 years.
- The difficult course of cirrhosis with complications is not more than 3 years.
The average indicators indicate that the primary and secondary shape of the cirrhosis ends with a fatal outcome for 8 years after the first symptoms appear. But it is extremely difficult to put accurate life expectancy forecasts, especially with the autoimmune development of the disease.
Anna, 29 years old writes: "The diagnosis was put 3 years ago, I had to go through many surveys. But the doctor reassured me that the disease revealed early stageAnd with timely treatment it is possible to suspend the disease. "
Bilyary cirrhosis is not only the most rare, but also the most dangerous of all kinds of disease. It is especially difficult to predict the development of primary cirrhosis, as well as choose treatment or undertake preventive measures. Patients with biliary lesions of the liver are important not to lower the hands, and adhere to the advice and appointments of the attending physician - with the right approach, it is possible to extend the length of life for several decades.