Pärilik kuulmiskahjustus vastsündinutel. Pärilik kuulmislangus ja kurtus. Ülevaade Pärilikud kuulmis- ja kõnepatoloogiad
kromosoomid. Ameerika teadlased K. Grader ja E. Blackburn viisid läbi uuringu, et selgitada välja, kas telomeer -DNA moodustamisel osaleb mõni varem tundmatu ensüüm. 1984. aasta jõulupühal avastas K. Greider rakuekstraktis ensümaatilise aktiivsuse märke. Avastatud ensüümi nimetasid E. Blackburn ja K. Greider telomeraas. Pärast selle eraldamist ja puhastamist leidsid teadlased, et see ei koosne mitte ainult proteiinist, vaid ka RNA -st, mis sisaldab sama järjestust nagu telomeer. Seega toimib RNA mallina telomeeride konstrueerimiseks, samas kui ensüümi valgukomponent on vajalik otse ensümaatiliseks aktiivsuseks. Telomeraas pikendab telomeeri DNA -d, pakkudes platvormi, mis omakorda võimaldab DNA polümeraasidel kopeerida kromosoomi kogu pikkuses ilma geneetilist teavet kaotamata. Seega ei lühendata kromosoomi kopeerimisel.
E. A. Tšistjakova
Nägemise ja kuulmise pärilikud kõrvalekalded - geneetiline alus, ravi ja ennetamine
Inimeste tervise ja geneetika probleem on omavahel tihedalt seotud. Geeniteadlased püüavad vastata küsimusele, miks mõned inimesed on vastuvõtlikud mitmesugused haigused, samas kui teised jäävad samadel tingimustel terveks. See on peamiselt tingitud iga inimese pärilikkusest, see tähendab tema geenide omadustest, mis asuvad kromosoomides.
Pärilikkus on inimkonna ajaloos alati olnud üks kõige raskemini seletatav. Isegi iidsetel aegadel kasutasid inimesed alateadlikult geneetilisi meetodeid taimede ja loomade aretamisel ning seoses inimestega oli eluvaatlusi, mis olid seotud erinevate tunnuste pärimisega: juuste värv, silmad, kõrva kuju, nina, huuled, kõrgus, füüsis, deformatsioonide pärimine ühe perekonna esindajatel.
Paljud teadlased on esitanud oma hüpoteesid pärilike patoloogiate esinemise kohta. Kuid nende eeldused ei põhinenud teaduslikel vaatlustel. XX sajandil. "geneetika" teaduse arenguga selgitati ja teaduslikult kinnitati, et sellised patoloogiad on päriliku iseloomuga. Pärilike haiguste uurimine on seotud teadusega, mida nimetatakse "meditsiiniliseks geneetikaks".
Selle teema asjakohasus on tingitud ebapiisavatest teadmistest nägemise ja kuulmise kaasasündinud anomaaliate etioloogiast (haiguste alguse põhjused ja tingimused) ning patogeneesist (haiguste tekkimise ja arengu mehhanism). Nendel haigustel on erinevaid vorme. Heidame kiire pilgu igaühele neist.
1. Pärilikud kõrvalekalded koos nägemise ja kuulmise halvenemise või kurtide pimedusega - kaasasündinud või omandatud varajane iga(enne kõne valdamist) pimedus ja kurtus, samuti kuulmispuudega seotud tumm. Ilma eriväljaõppeta ei arene kurt-pime-tumm laps vaimselt, ei omanda elementaarseid enesehooldusoskusi. Seetõttu realiseeritakse kurt-pimedate jaoks arenguvõimalus erihariduse protsessis.
Kõige tavalisemad pärilike kombineeritud nägemis- ja kuulmisanomaaliate sündroomid: Usher, Marshall, Reetur Collis, Cruson, Alport.
2. Nägemispuudega kuulmise pärilikud kõrvalekalded.
Välis- ja keskkõrva kaasasündinud väärarendid lapsepõlv on üsna levinud. Meie riigis sünnib aastas keskmiselt kuni 400-500 selle patoloogiaga last. Nende hulgas erinevad tüübid laste ENT-patoloogia, kõrva kaasasündinud väärarengud moodustavad ligikaudu 5-6%.
Kurtide lapse vaimne areng on erinev sõltuvalt sellest, kas tema kurtus on kaasasündinud, kas ta kaotas kuulmise ontogeneesi varases staadiumis. Sensoorse sfääri häirete taksonoomia arvestab aja ja kahjustuse astme kriteeriume. Kuulmispuudega laste hulgas paistavad silma järgmised:
1) kurt: a) kurt varakult; b) hiline kurt;
2) kuulmispuudega: a) suhteliselt terve kõnega; b) sügava kõnearenguga.
Haigused, mille puhul kuulmise arengus esineb kõrvalekaldeid: välise kuulmiskanali atreesiaga makrotoonia, Pendred, Richards-Randal, Jervell ja Lange-Nielsen, Waa-denburgi sündroomid.
3. Visuaalsed kõrvalekalded ilma kuulmispuudega.
Mõned nägemishäired on sarnased kuulmispuudega. Päriliku haiguse korral ei saa laps teatud kogust visuaalseid kujutisi. Raskused tekivad "tekkimisel" vertikaalne asend kehad, hirm kosmose ja uute objektide ees võivad põhjustada ruumi arengu ja teatud tegevuse hilinemise. Esimesed manipulatsioonid ja individuaalsed toimingud objektiga ilmuvad pimedatele lastele alles kahe aasta pärast.
Nägemiskahjustus varases eas, isegi halva nägemise korral, põhjustab ka psühhomotoorse sfääri vähearenenud arengut: täheldatakse haardumistegevuse nõrkust, liigutuste diferentseerumist viivitatakse, täheldatakse külmumist või vastupidi - liigseid stereotüüpseid liigutusi. pea ja käed.
Vanemas eas nägemise kaotanud lapse areng areneb aga hoopis teistmoodi. Olemasolev visuaalsete muljete varasem kogemus hõlbustab motoorsete oskuste arendamist, objektide tegevust, ideede ja kontseptsioonide kujundamist.
Nägemispuudega laste hulgas on:
a) täiesti pime;
b) osaliselt nägemine;
c) nägemispuudega.
Nägemisorgani avastatud kaasasündinud patoloogia esinemissagedus on 2-4%. Geneetilised muutused põhjustavad 50% laste pimedusest.
Kõige tavalisemad pärilikud haigused, mis põhjustavad nägemiskahjustusi või pimedust: Riegeri, Alstromi, Lenz, Aperti, Marfani sündroomid.
Seega näeme, et päriliku eelsoodumusega haigused on suur mitmekesine haiguste rühm, mille areng on tingitud teatud pärilike tegurite (näiteks mutatsioonide) koosmõjust. Päriliku eelsoodumuse aluseks on inimpopulatsiooni lai polümorfism ensüümides, struktuur- ja transpordivalkudes, antigeenides, mis tagab iga inimese geneetilise unikaalsuse.
Tänu meditsiinigeneetika edusammudele ja ideede laienemisele igasuguste haiguste pärimise olemuse kohta on mutantsete geenide avaldumine, raviviisid muutunud palju selgemaks ja mis kõige tähtsam - pärilike haiguste ennetamine on kõige olulisem. meditsiinigeneetika oluline ülesanne. See võimaldab teil õigeaegselt vältida haigete laste sündi. Pärilike haiguste ennetamine toimub peamiselt meditsiiniliste geneetiliste konsultatsioonide ja diagnostikakeskuste kaudu.
Pärilike haiguste ravi üldised lähenemisviisid on sarnased mis tahes muu etioloogiaga haiguste raviga. Päriliku patoloogia ravis säilib individuaalse ravi põhimõte. See põhimõte on eriti oluline, kuna pärilikud haigused on heterogeensed ja samasugused kliiniline pilt võivad esineda erinevad pärilikud haigused, millel on erinev patogenees. Sõltuvalt genotüübist, sünnieelse ja -järgse ontogeneesi tingimustest, mutatsioonide avaldumisest
konkreetset isikut saab muuta. Pärilike haiguste ja päriliku eelsoodumusega haiguste ravis eristatakse järgmisi valdkondi:
Sümptomaatiline (mõju sümptomitele);
Kirurgiline (elundite ja kudede korrigeerimine, eemaldamine või siirdamine);
Patogeneetiline (mõju kehas esinevatele protsessidele haiguste ajal);
Etioloogiline (või etiotroopne - ravi, mille eesmärk on kõrvaldada haiguse põhjus).
Päriliku patoloogia ennetamiseks on teatud tüüpi.
1. Esmane ennetus hõlmab tegevusi, mis takistavad haige lapse sündi. See saavutatakse sünnituse planeerimisega, valides optimaalse reproduktiivse vanuse, keeldudes last kandmast päriliku ja kaasasündinud patoloogia kõrge riski korral. Umbes 20% kõigist põlvkondade pärilikest haigustest on haigused, mis on põhjustatud uutest mutatsioonidest.
2. Sekundaarne ennetus viiakse läbi raseduse katkestamisega, kui on suur tõenäosus lootehaiguste või sünnieelselt diagnoositud haiguste tekkeks. Raseduse katkestamine on praegu ainus praktiliselt teostatav meetod kõige raskemate ja surmavate geneetiliste defektide ennetamiseks.
3. Päriliku patoloogia kolmanda astme ennetamine tähendab patoloogiliste genotüüpide ilmingu korrigeerimist. Tema abiga on võimalik saavutada täielik normaliseerumine või patoloogilise protsessi raskusastme vähenemine. Päriliku haiguse arengu ennetamine hõlmab terapeutiliste meetmete kogumit, mida saab teha emakas või pärast sündi. Sellisel juhul on ennetusmeetmed tihedalt seotud pärilike haiguste raviga ja nende vahel pole selget piiri.
Pärilik kuulmislangus - kuulmiskahjustus päritud perekonnas, kus on juhtumeid eelmistel põlvkondadel kuulmislangus... Põhjus on muutunud geenide pärimine. Tõenäoliselt teate juba, et geenid on päriliku teabe kandjad, mis määravad kõigi elusolendite arengu. Kõik geenid kannavad kogu organismi arenguks vajalikku teavet ühest viljastatud munarakust. Iga geen vastutab individuaalselt konkreetse valgu moodustumise eest. Valgud ehitavad kogu keha. Me kõik saame igast geenist kaks eksemplari: üks emalt, teine isalt. Seega on igal inimesel sama geeni kaks varianti. Kuulmisorgani moodustamise ja toimimise eest vastutavad mitmed keha geenid. Kokku on selliseid geene vähemalt 100. Pole üllatav, et hiljutiste uuringute kohaselt on üle 50% kõigist kaasasündinud ja varases lapsepõlves kuulmislanguse juhtumitest seotud pärilike põhjustega. Arvatakse, et igal kaheksandal Maa elanikul on üks geenidest, mis põhjustavad retsessiivset kuulmislangust.
Mis on päriliku kuulmislanguse kõige levinum tüüp?
Ligikaudu 75% kõigist päriliku kuulmislanguse juhtudest on tingitud retsessiivne mittesündroomne kuulmiskahjustus (RHND) või retsessiivne mittesündroomne kuulmislangus.
Retsessiivse päranditüübi korral saab laps igalt vanemalt sama geenivariandi, mis põhjustab seda kuulmiskahjustust. "Retessiivne" geen avaldub ainult koos teise sama geeniga ja põhjustab RNNS -i. Samal ajal ei kannata lapse vanemad kuulmispuudega, kuna neil on selle geeni normaalne variant vanematelt saadud geenipaaris. Siiski on nad geeni kandjad retsessiivne mittesündroomne kurtus... Seega võib lapsel olla kuulmispuue, samas kui tema vanematel ja kõigil teistel sugulastel võib normaalne kuulmine olla igas vanuses.
Mittesündroomse vormi all mõistetakse asjaolu, et kuulmiskaotusega ei kaasne muid tunnuseid või teiste elundite ja süsteemide haigusi, mis päriksid koos sündroomivormides esineva kuulmislangusega. Näiteks Pendredi sündroom - kuulmislanguse kõige levinum sündroomne variant - on retsessiivne haigus, mida iseloomustab kuulmiskahjustuse ja eutüreoidse struuma moodustumise kombinatsioon. Goiter areneb noorukieas ja hiljem, nii lastel diferentsiaaldiagnostika Pendredi sündroom ja mittesündroomne retsessiivne kuulmislangus on äärmiselt rasked.
Autosomaalsed domineerivad vormid mittesündroomne kuulmislangus on suhteliselt haruldane ja moodustab mitte rohkem kui 25% kõigist mittesündroomilistest kurtustest. Domineerivates vormides piisab haiguse avaldumiseks ühest muudetud geenikoopiast. Sellistel juhtudel on reeglina üks vanematest kuulmislangusega lapsel haige, kuid võimalik on ka uus mutatsioon.
Kuidas laps RNNS -i saab?
Niisiis, igaüks meist saab pooled geenid oma isalt ja teise poole emalt. Milline geen vanemate geenipaarist saame, on puhtalt juhuslik nähtus.
Aeg -ajalt võib kokkupuude mis tahes teguriga põhjustada geenimuutuse. Geneetikud nimetavad seda muutust mutatsiooniks. Paljud inimesed ei tea, et nad on muutunud geenide kandjad. Need muutused, kui need on ilmnenud, päritakse põlvest põlve. Enamik mutatsioone ei mõjuta keha seisundit, kuid mõnikord avaldavad mõned neist oma mõju mitmel põhjusel. Üks põhjusi on sama muudetud geeni kahe kandja kohtumine. Nad võivad olla retsessiivse mittesündroomse kurtusega lapse vanemad. See laps saab muudetud geeni igalt vanemalt ja tal on seega kaks muudetud geeni koopiat. Ainult sel juhul avaldab mutatsioon oma mõju geeni normaalse variandi puudumise tõttu. Nende vanemate puhul on kaasasündinud mittesündroomse kurtusega lapse saamise oht 25%. Tavaliselt on kuulmislanguse aste esialgu korrigeerimiseks ja treenimiseks piisav. Nimetatud abielus normaalse kuulmisega lapsed võivad sündida 75% juhtudest ning mõnel lapsel võib olla tervislik genotüüp (25%) ja miski ei ohusta neid tulevikus, samas kui teised, nagu nende vanemad, on muutunud geen (50%) ja nende jaoks võib olukord korduda.
Kahe kuulva vanema, muutunud geeni kandjate abielu
Kuulmiskaotuse arengu jaoks oli kõige olulisem konnektiin-26 geen (GJB2)... Ainult üks selle geeni muutus, mida nimetatakse 35delG mutatsiooniks, põhjustab 51% kõigist kaasasündinud ja varase lapseea kuulmiskaotuse juhtudest. Selles geenis on ka teisi teadaolevaid muutusi. Tänu läbiviidud uuringutele on teada, et meie riigis on iga 20 elanikku 35delG mutatsiooni kandja. Seetõttu on kahjuks muutunud geeni kandjatega kohtumise tõenäosus üsna suur.
Millist kasu oodatakse RNNS -i viivate geenide tuvastamisest?
See on võime täpselt välja selgitada kuulmispuude põhjused. Geneetiline analüüs võimaldab teha õige prognoosi haiguse kordumise kohta perekonnas, samuti hinnata kuulmispuudega laste saamise tõenäosust sugulaste peredes. Geneetilise defekti varajane avastamine aitab õigeaegselt valida õige taktika kuulmispuudega lapse raviks ja rehabilitatsiooniks ning säästab teda tarbetute ravimite võtmisest.
Isegi terve inimene saab teada oma genotüübi, kuna ta võib olla kuulmislanguse geeni kandja (tõenäosusega 1/20). Kuulmislanguse geeni kandmise uuringud on eriti olulised inimestele, kellel on kuulmispuudega sugulasi, samuti abikaasadele, kes on tihedalt seotud abielus.
Abielupaaril, kus mõlemad on kuulmislanguse geeni mutatsiooni kandjad (olenemata sellest, kas neil on juba kuulmispuudega laps või mitte), on võimalik haiguse sünnieelne (sünnieelne) DNA -diagnostika läbi viia loode raseduse varases staadiumis (9-12 nädalat).
Kuidas toimub RNNS -i viivate geenide otsimine?
Rohkem kui pooled kaasasündinud RNNS -i juhtudest on tingitud geeni homosügootsetest ja heterosügootsetest retsessiivsetest mutatsioonidest GJB2 (geneetilise tüübi DFNB1 kuulmislangus: OMIM 220290 ). Selle kuulmislanguse geneetilise tüübi sagedus on 1: 1000 vastsündinut. Mõned haruldased spetsiifilised mutatsioonid geenis GJB2 näitavad domineerivat-negatiivset mõju ja põhjustavad heterosügootses seisundis autosomaalse domineeriva mittesündroomse kuulmislanguse (DFNA3A: OMIM) 601544 ) või sündroomid keratiit-ihtüoos-kurtus (OMIM 148210 ), Fovinkel (OMIM 124500 ), Bartha Pumphrey (OMIM 149200 ), palmar-plantaarne keratoderma koos kurtusega (OMIM 148350) ... Molekulaargeneetika keskuses tehakse kindlaks, kas patsiendil on geenis mutatsioone GJB2... See uuring sisaldab otsige 8 kõige levinumat geeni mutatsiooniGJB2 (c.35delG, c.-23 + 1G> A (IVS1 + 1G> A), c.101T> C (p.Met34Thr), c.313_326del14, c.235delC, c.167delT, c.358_360delGAG (lk. Glu120del) ja del (GJB2-D13S175)), moodustades 95% geeni mutatsiooniga kromosoomide koguarvust GJB2, alleelispetsiifilise MLPA järgi; ja geeni mutatsioonide otsimineGJB2 järjestamise teel mRNA järjestused (eksonid 1 ja 2) ja eksoni-introni geenide ristmikud. Rakendatud lähenemisviis võimaldab tuvastada rohkem kui 99% geeni mutatsioonidest GJB2... Vajadusel teostatakse otsige pikka kustutamist 309 kb del (del ( GJB6) -D13S1830) lookusesDFBN1.
HNS -iga patsientidel ilma geenimutatsioonideta GJB2 mutatsioone täheldatakse teistes geenides: HHC eest vastutavaid geene on kirjeldatud rohkem kui 100. Molekulaargeneetika keskus otsib 32 kõige tavalisema HNS -i geneetilise vormi mutatsioone ja sündroome, mis maskeeruvad sellistena patsientidel, kellel ei ole geeni mutatsioone. GJB2 meetod 32 geeni sekveneerimispaneel tabelis esitatud.
|
Haigus |
||
|
DFNB16, C. kurtus ja meeste viljatus |
||
|
Ushera küla 1B,DFNB2,DFNA11 |
276900, 600060, 601317 |
|
|
DFNB21, DFNA8 / 12 |
||
|
DFNB4, umbes Pendreda |
||
|
DFNB12, lk. Ushera 1D |
||
|
Ushera küla 2A |
||
|
DFNB8 / 10 |
||
|
DFNB7 / 11, DFNA36 |
||
|
DFNB53,DFNA13, C. Kleebise tüüpIII, lk. Nancy-Sweeney-Insley sündroom |
609706, 601868, 184840, 215150 |
|
|
DFNB23, lk. Ushera 1F |
||
|
DFNA6 /DFNA14, S. Wolfram |
600965, 222300, 614296 |
|
|
koos. Usherah 2C |
||
|
DFNB37,DFNA22 |
||
|
DFNA20 / 26, lk. Baraitser-Winter tüüp 2 |
||
|
DFNB84, DFNA73 |
||
|
DFNA17, lk. Alport koos leukotsüütide ja makrotrombotsütopeeniaga |
||
|
DFNB59 /PJVK |
||
|
DFNB31, lk. Usherah 2D |
||
|
Alstrom s. |
||
Sünnieelse (sünnieelse) DNA diagnostika läbiviimisel konkreetse haiguse puhul on mõistlik diagnoosida sagedased aneuploidiad (Downi, Edwardsi, Shereshevsky-Turneri sündroomid jne) juba olemasoleval loote materjalil, punkt 54.1. Selle uuringu asjakohasus on tingitud aneuploidiate suurest üldsagedusest - umbes 1 300 vastsündinu kohta ja loote materjali uuesti proovide võtmise puudumisest.
Universaalne vastsündinute sõeluuringute programmid, mis hõlmab kõikide vastsündinute kuulmisfunktsiooni elektrofüsioloogilist testimist enne haiglast väljakirjutamist, on kogu Ameerika Ühendriikides seaduslikult vastu võetud või vabatahtlikult vastu võetud. Ja kuigi need programmid on võimaldanud paljudel, paljudel lastel varakult avastada kuulmiskaotust, ei suuda nad kunagi tuvastada absoluutselt kõiki kaasasündinud kuulmiskaotuse juhtumeid, kuna väga sageli ei hakka geneetiliselt määratud kuulmislangus ilmnema kohe pärast sündi. .
Vara kuulmislanguse diagnostika võimaldab läbi viia varasemaid terapeutilisi meetmeid (logopeedia, kuuldeaparaadid, kõrvaklapi siirdamine), mis lihtsustavad kõne- ja keeleoskuse omandamist ning parandavad õpitulemusi.
Arenenud riikide pärilikud vormid moodustavad umbes 60% kõigist kuulmislanguse vormidest. On tuvastatud üle 400 geeni, mis vastutavad kuulmislanguse arengu eest. Kõigist pärilikest kuulmislanguse vormidest on umbes 70–80% mittesündroomilised vormid (ülejäänud 20–30% on sündroomivormid).
Tähtaeg kaasasündinud kuulmislangus tähendab, et kuulmine on alates sünnist halvenenud, näiteks kaasasündinud tsütomegaloviiruse infektsiooni korral. Pärilik kuulmislangus võib avalduda sündides ja pärast seda, mõnel juhul isegi paljude aastate pärast. Kuulmiskaotus võib olla ühepoolne ja kahepoolne, progresseeruv ja mitte progresseeruv. Erinevate geneetiliste defektidega kuulmislanguse aste ja olemus on erinevad. Mõnikord võivad need varieeruda isegi sama geneetilise haiguse raames.
a) Mendeli kuulmispuude pärimine (kurtus)... Mendeli päranditüüpide hulka kuuluvad autosomaalne dominant (AD), autosomaalne retsessiivne (AR), X-seotud retsessiivne ja X-seotud dominant. Autosomaalse domineeriva pärandi puhul on sageli üks abikaasa (haige) heterosügootne (üks geeni alleel on muutunud, teine mitte) ja teine abikaasa (terve) on homosügootne (mõlemad alleelid ei muutu).
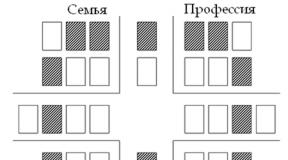
Kui me vaatame selle olukorra nn Pennetti võre peal näeme, et kuulmislangusega järeltulija sündimise tõenäosus on 50%. Järglased, kes ei päri Dd genotüüpi, ei kanna kuulmislanguse tekkimise eest vastutavat alleeli. Perekonna ajalugu hinnates leitakse tavaliselt, et geen edastatakse vertikaalselt, s.t. geen avaldub mitmel järjestikusel põlvkonnal.
Kell autosomaalne domineeriv pärand sugu ei ole enamikul juhtudel oluline, seetõttu võib kuulmislangus mõlema soo vanematelt sama tõenäosusega edasi kanduda. Kui ehitada Pannetti võre kahele haigele Dd genotüübiga lapsevanemale, näeme, et haige homosügootse DD järglase tõenäosus on 25%, haige heterosügootse Dd järglase tõenäosus 50%ja terve homosügootse dd tõenäosus järglasi on 25%. DD genotüübiga haige inimese fenotüüp on tavaliselt raskem kui heterosügootsete Dd järglaste fenotüüp. Dd terved, homosügootsed järglased ei edasta haigust oma lastele.
Kui köidetakse teatud tingimuste genotüübiga(näiteks Dd) ei kaasne selle manifestatsiooniga (näiteks kuulmislangus), arvatakse, et genotüübil on puudulik läbitungivus. Ekspressiivsus viitab fenotüüpide varieeruvusele, mida saab seostada ühe genotüübiga (nt Dd); seega võib kahel sama genotüübiga inimesel olla erinev fenotüüp.
Kell autosoomne retsessiivne pärand tavaliselt on mõlemad vanemad terved heterosügootid (üks muudetud alleel ja üks normaalne alleel). Sel juhul näeme Pannetti võret vaadates, et 25% tõenäosusega areneb lapsel kuulmislangus ja 50% tõenäosusega on ta kandja. Perekonna ajalugu hinnates selgub tavaliselt, et kuulmislangus edastatakse "horisontaalselt", s.t. esineb mitmel sama põlvkonna liikmel, kuid mitte mitmel järjestikusel põlvkonnal. Nagu autosomaalse domineeriva pärandi puhul, pole ka sool enamikul juhtudel tähtsust, seetõttu võib kuulmislangus mõlema soo vanematelt võrdse tõenäosusega edasi kanduda.
Kui abielu sõlmitud haigete vanemate vahel, kes on retsessiivse tunnuse (rr) suhtes homosügootsed, mis tähendab, et kuulmislangusega järeltulija sündimise tõenäosus on 100%.
Kell X-seotud retsessiivne pärand haigus avaldub fenotüübis meestel, kellel on muutunud alleel X -kromosoomis, ja naistel, kellel on muutunud alleeli kaks koopiat. Kõige sagedasem ülekanne on naissoost kandja ja terve isa. X-seotud retsessiivse pärandi jaoks ehitatud Pannetti tabeli kohaselt on sellise kandja tüdruku saamise tõenäosus 50%, haige poisi saamise tõenäosus samuti 50%. Haige mees ei anna seda haigust kunagi edasi oma poegadele; poiss saab isalt terve Y -kromosoomi ja emalt X -kromosoomi.
Vedaja naine võib edastada muutunud alleeli nii tütrele kui ka pojale. Haige isase (xY) ja emase kandja (xX) juuresolekul on tõenäosus, et poiss kannatab päriliku kuulmislanguse all, 50%, tüdrukul on pärilik kuulmislangus 50% (xx) , tõenäosus, et tüdruk on vedaja, on samuti 50%.
X-seotud domineeriv pärand on palju vähem levinud kui X-seotud retsessiivne. X-seotud domineeriva pärandi korral sõlmitakse abielu tavaliselt xX genotüübiga haige naise ja XY genotüübiga terve mehe vahel. Nagu autosomaalse domineeriva pärandi puhul, ilmneb haigus xX genotüübi juuresolekul fenotüübis. Allolevas tabelis on kujutatud Pannetti võre haige naise (tavaliselt heterosügoot) ja terve mehe jaoks. Tõenäosus, et naissoost järglased kannatavad kuulmislanguse all, on 50% (genotüüp xX) ja tõenäosus, et meessoost järglased kannatavad kuulmispuude all, on samuti 50% (genotüüp xY). Mõelge abieluolukorrale terve naise (XX) ja haige mehe (xY) vahel.
Sel juhul 100% naissoost järeltulijad on kurt, sest nad saavad isalt muudetud domineeriva alleeli x, kuid samal ajal on kõik 100% isased järglased terved, sest nad saavad emalt modifitseerimata X alleeli.
Kell X-seotud domineeriv pärand fenotüüp on meestel rohkem väljendunud kui naistel. Naistel, kellel on xX genotüüp, osutub üks alleel normaalseks, samas kui meestel on X -kromosoomis ainult üks muudetud alleel, mille tagajärjel hakkab muudetud geen patoloogilist valku sünteesima (või selle sünteesi häirima).
Keelt parandades kurtidele ja tummadele ning kurtide koolide tekkimisele, kurtidel on võimalus üksteisega tihedamalt suhelda, mille tulemusel on suurenenud nendevaheliste abielude arv (assortatiivne ristumine). Päriliku retsessiivse kuulmislanguse all kannatavad patsiendid on sama muutunud alleeli homosügootid. Järelikult on 100% nende järglastest kurt. Selliseid abielu, kus 100% järglastest on kurt, nimetatakse mitte-täiendavateks. "Täiendav" on abielu, milles kõik vanemate järeltulijad erinevaid vorme pärilik kurtus (kas omandatud kurtusega vanemate vahel või ühe omandatud kurtusega vanema ja teise autosoomse retsessiivse kurtusega) on normaalse kuulmisega.

b) Kuulmiskahjustuse (kuulmislangus) mitokondriaalne pärimine... DNA -d leidub mitte ainult rakutuumades, vaid ka tsütoplasma mitokondrites. Mitokondrid varustavad rakke energiaga adenosiintrifosfaadi (ATP) kujul ja osalevad ka teistes rakuprotsessides (rakkude diferentseerumine, apoptoos, signaali edastamine). Iga rakk sisaldab palju mitokondreid. Kui rakk sisaldab muutunud ja normaalse DNA -ga mitokondreid, nimetatakse seda seisundit mitokondriaalse DNA heteroplasmiaks. Kui rakk sisaldab ainult muudetud DNA -ga mitokondreid, nimetatakse seda seisundit mitokondriaalse DNA homoplasmiaks. Homoplasmia korral on sümptomid tavaliselt raskemad ja ilmnevad varem kui heteroplasmia korral. Heteroplasma on tavalisem kui homoplasma; Kuna muutunud mitokondrite arv võib olla erinev, on heteroplasmias fenotüübi ekspressiooni varieeruvus suurem kui homoplasmias.
Mitokondriaalne DNA päritakse ainult emalt, mitte isalt, sest mitokondriaalset DNA -d leidub munades, kuid mitte spermas. Mitokondriaalne kuulmislangus moodustab umbes 1% kõigist keelealuse kuulmiskaotuse juhtudest ja 5-10% kõigist postlingvaalse, mittesündroomse kuulmislanguse juhtudest.
v) Mittesündroomne kuulmislangus... Enamikul juhtudel (70-80%) on pärilik kuulmislangus mittesündroomne. Neist umbes 65-75% on päritud autosoomse retsessiivse viisil. Iga autosomaalse retsessiivse mittesündroomse kuulmislangusega seotud geeni lookus on tähistatud DFNB ja araabia numbriga.
Kuulmiskaotus, mis on seotud konnektiini valkude kahjustusega... Kõige sagedamini on autosomaalsel retsessiivsel viisil päritud mittesündroomne kuulmislangus seotud mutatsioonidega sidekoeproteiinide perekonnas, eriti vaheühenduse valgu (36, vaheühenduse valk) geenis GJB2, mis kodeerib konnektiini 30. Mutatsioonid GJB2 geen võib põhjustada kuni 50 % kõigist tõsise mittesündroomse kuulmislanguse juhtudest, mis on päritud autosomaalselt retsessiivsel viisil sisekõrva karvarakud.
Kõige sagedasem mutatsioon konnektiinide perekonnast on GJB2 mutatsioon, kõige levinum konnektsiin 26 mutatsioon on 35delG / 30delG (kõige levinum eurooplastel ja valgetel ameeriklastel). 167delT mutatsiooni leidub kõige sagedamini Aškenazi juutidel, 235delC aasialastel, R143W mõnel Aafrika populatsioonil, W24X hispaanlastel, slovakkidel ja mõnel indiaanlasel. Siiani on tuvastatud umbes 90 erinevat GJB2 mutatsiooni. Kuigi enamik neist on seotud mittesündroomse autosomaalse retsessiivse kuulmislangusega, on mõned GJB2 mutatsioonid seotud kuulmiskaotuse sündroomivormidega, mis päritakse autosomaalselt domineerival viisil (näiteks Fowinkeli sündroom või ektodermaalne düsplaasia keratiidi-ihtüoosi korral). kuulmislanguse sündroom).
Kaasasündinud ja omandatud püsivad kuulmiskahjustused
- Haiguse struktuuri analüüs.
Erinevate kirjandusallikate andmetel kannatab 4–6% meie planeedist ühe või teise kuulmispuude all. Oletame vastavalt akadeemik N. A. Preobraženskile, et kurtide ja vaegkuuljate koguarv on 5%. Kui me loeme sada, on Maa elanikkond jõudnud juba 5 miljardini. inimesi, siis on igas vanuses kuulmispuudega inimeste arv 250 miljonit.See on lähedane sellise riigi elanikkonnale nagu USA. Arvestades sellise seisundi nagu kuulmislangus sotsiaalpsühholoogilist olemust, on see probleem kahtlemata lapsepõlves eriti oluline. Kodused ja paljud välismaised kõrva -kurguarstid pööravad tähelepanu erinevate kuulmiskahjustuste leviku ja olemuse väljendunud sõltuvusele laste vanusest. Anamneesi analüüs võimaldas tuvastada, et enamik lapsi - 82% - kannatab kuulmisorgani kahel esimesel eluaastal, s.t. enne kõne arengut ja selle kujunemise ajal.
Kuulmisorganite haiguste struktuuri olemasolul vastavalt Venemaa piirkondadele aastatel 1991-1992. domineeris kuulmislanguse sensorineuraalne vorm (72,6%), juhtiv kuulmislangus diagnoositi 13,3%, sega - 14,1%patsientidest. Järgnevatel aastatel muutus pilt juhtiva ja segaste kuulmislangusega laste arvu suurenemise tõttu. Püsiv kuulmiskahjustus - kuulmisfunktsiooni kahjustus, mis ei näita märkimisväärset paranemist nii iseseisvalt kui ka ravi tulemusena, s.t. on pöördumatud.
Lapsepõlves püsiva kuulmiskahjustuse põhjused võib jagada kolme rühma: pärilikud, kaasasündinud ja omandatud.
- Pärilikud häired.
Geneetilised kuulmishäired avalduvad kurtuses ja kuulmislanguses. Pärilik kuulmislangus on sensineuraalne, mida iseloomustavad pöördumatud muutused kuulmissüsteemi struktuurides. Tema tavalised omadused on järgmised:
Helitaju kahepoolne kahjustus;
Kaasamine Corti elundi patoloogilisse protsessi;
Vestibulaarsete häirete puudumine.
Kurtus on enamasti pärilik. 37%-l edastatakse kurtus retsessiivsel viisil, 12%-l domineerival viisil, 2%-l on kurtus seotud sooga.
Pärilik kurtus on jagatud kahte kategooriasse:
Kuulmiskahjustus kui monosümptom;
Kuulmiskahjustus kui sekundaarne sündroom erinevate elundite ja süsteemide kahjustuste kompleksis.
Esimesel juhul eristatakse järgmisi morfoloogilisi muutusi kuulmissüsteemis, sõltuvalt nende levimusest ja lokaliseerimisest:
Puudumine sisekõrv ja vahel kivine osa ajaline luu normaalselt arenenud välis- ja keskkõrvadega;
Domineeriva kurtusega täheldatakse mitmesugust labürindi luu- ja membraaniosade vähearenenud arengut (kõrvade lokkide arv väheneb, vestibulaarne labürint on vähearenenud, kott on laienenud);
Kõrvaääre sensoorsete struktuuride vähearenenud;
Degeneratiivsed häired spiraalganglioni rakkudes ja sisekõrva närvi kiududes.
Teist päriliku kurtuse kategooriat täheldatakse paljude pärilike haiguste korral, mis on põhjustatud keha väliskihtide, luu-, närvi-, endokriinsüsteemid, siseorganite haigused.
Isegi leopardi sündroomiga (tedretähnide ilmumine kohe pärast sündi) võib esineda kurtus.
Mõned pärilikud kuulmisvead on progresseeruvad. Mõnikord on need kombineeritud teiste defektidega: nägemise halvenemine, luure, neeruhaigus, lihasluukonna, naha- ja muud häired. Sisekõrva pärilikud häired arenevad sageli koos välise ja keskkõrva kõrvalekalletega.
Praegu eristan üle 60 päriliku kurtuse tüübi koos sisekõrva kahjustusega. Kõiki neid saab rühmitada järgmiselt:
Micheli tüüp (labürindi või idapoolse luu püramiidi osa puudumine välis- ja keskkõrva normaalse arenguga);
Tüüp Mondini, Shaibe, Alexander (mitmesugused vead labürindi arengus);
Degeneratiivsed muutused spiraalse sõlme rakkudes ja kõrvaklapi närvi kiududes, mis avalduvad täiskasvanueas.
Alporti sündroom: punaste vereliblede, leukotsüütide ja valkude ilmumine uriinis.
Alstromi sündroom: raske pärilik haigus, mis päritakse autosomaalselt retsessiivsel viisil.
Cockayne'i sündroom: pärilik haigus, mis edastatakse autosomaalselt retsessiivsel viisil. Kahe aasta pärast on vaimse ja füüsilise arengu mahajäämus.
- Kaasasündinud häired
Endo- või eksogeensete patoloogiliste mõjudega loote kuulmisorganile sünnituse ajal või vastsündinute perioodil võib päriliku koormatud tausta puudumisel tekkida kurtus või kuulmislangus, mida nimetatakse kaasasündinud. Kuulmisfunktsiooni kaasasündinud häired on tavalised ja avalduvad nii heli tajumise kui ka heli juhtimise patoloogias, sagedamini on neil sensorineuraalse kuulmislanguse iseloom.
Loote kuulmisfunktsiooni mõjutavate põhjuste hulgas on kõige sagedasemad raseduse 1., 2. poole toksikoos ja emakasisesed infektsioonid: eriti punetised, mille ema kandis üle raseduse esimesel trimestril. Ja veel: leetrid, gripp, viirushepatiit, tuulerõuged, epideemiline paratitis, tsütomegaalia, toksoplasmoos, kaasasündinud süüfilis jne.
Olulist rolli kuulmispuude tekkimisel mängib vastsündinute kernicterus, mis on põhjustatud ema ja lapse konfliktist Rh -faktori või veregrupi üle. Arvatakse, et kuulmisnärvid on eriti vastuvõtlikud bilirubiini toksilisusele, mis esineb vastsündinute kernikteruses.
On tõestatud, et enneaegsetel imikutel esineb kuulmispuude esinemissageduse suurenemine, eriti ema ja loote vere kokkusobimatuse tõttu Rh -teguri ja veregrupi kokkusobimatusega nende laste sugupuu kuulmisvigade esinemisel. kombinatsioon enneaegsusest erinevate kaasasündinud väärarengutega närvisüsteem ja muud organid.
Kuulmiskahjustus võib tekkida ka ema alkoholismi korral.
- Omandatud rikkumised
Omandatud häired esinevad mitmesuguste põhjustega, kõige tõsisemad heli vastuvõtva aparatuuri (sisekõrva, kuulmisnärv).
Laste kuulmispuude põhjuste hulgas on esikohal ägeda keskkõrvapõletiku tagajärjed. Kuulmislangus on kroonilise mädase keskkõrvapõletiku üks peamisi sümptomeid. Tavaline põhjus Kuulmislangus on nina ja ninaneelu haigused ja sellega seotud kuulmistoru obstruktsioon (adenoidid).
Pärast sündi võivad kuulmiskahjustused tekkida erinevate infektsioonide tõttu:
Hingamisteede haigused;
Gripp; leetrid; skarlotina, miningiit, epideemiline paratitis (mumps);
Tüsistunud keskkõrvapõletik või kuulmisnärvide toksilise neuriidi sümptomite tekitamine.
Märkimisväärne koht minengiidiga kuulmislanguse tekkes on hemetotsefaalsete ja hematolabüritiinbarjääride kaitseomaduste rikkumisel.
Epideemilise parasiidi korral areneb ühepoolne kurtus kiiresti koos vestibulaarse erutusvõime ühepoolse kadumisega. Gripi korral võib kuulmise muutus olla erinev - alates ühe kõrva täielikust äkilisest kurtusest kuni järkjärgulise aeglase languseni kuni täieliku kurtuseni erinevatel aegadel pärast haigust.
Nakkusliku hepatiidi korral on kuulmiskahjustus seotud joobeseisundist tingitud veresoonte seinte läbilaskvuse muutusega maksa võõrutusfunktsiooni nõrgenemise tagajärjel.
Sõltuvalt protsessi dünaamikast on kaks kuulmispuudega vormi:
Kiire, terav;
Aeglane, krooniline.
- Püsiva kuulmiskahjustuse klassifikatsioon
Puhtalt meditsiinilisest seisukohast. eriti olulised klassifitseerimise kriteeriumid on:
Kuulmiskahjustuse põhjused;
Protsessi lokaliseerimine;
Patoloogilise protsessi käik.
Alates psühholoogilisest ja pedagoogilisest tz. esile tulevad kriteeriumid, mis põhinevad lapse kõne arengut oluliselt mõjutavatel teguritel:
Kuulmiskahjustuse aste;
Kuulmispuude tekkimise aeg (sünnist või pärast kõne moodustumist);
Rikkumise alguse olemus (äkiline, järkjärguline).
Praegu on kuulmispuude kahe peamise kategooria - kurtus ja kuulmislangus - eristamise kriteeriumiks kuulmislanguse erinev aste.
Kurtus - püsiv kuulmislangus, mille puhul kõne sõltumatu valdamine ja kellegi teise kõne loetav tajumine on võimatu isegi kõige lähemal kõrvast. Samal ajal säilivad kuulmisjäägid, mis võimaldavad lähedalt tajuda valjuhäälseid helisid ja mõningaid kõnehelisid. See ei ole mitte ainult kuulmislangus üle 80 dB, vaid ka kuulmise kaotus või vähenemine erinevatel sagedustel, eriti kõnes.
Kuulmislangus - püsiv kuulmislangus, mille korral on võimalik allesjäänud kuulmisjääkide põhjal koguneda minimaalse kõnereservi järele, adresseeritud kõne tajumine vähemalt kõrvapulgast lähimal kaugusel. Kuulmislangus alla 80 dB.
- Kuulmiskaotuse klassifikatsioon
Kõik praktikas kasutatavad kuulmiskaotuse klassifikatsioonid põhinevad kuulmisteravuse kvantitatiivsel meetodil määramise põhimõttel (tabelid nr 8-9-10-11)
1.7. Kurtide klassifikatsioon
Rühm 1 - lapsed, kes tajuvad ainult madalaimaid sagedusi (128-256Hz);
Rühm 2 - lapsed, kes tajuvad madalaid sagedusi (kuni 512Hz);
Rühm 3 - lapsed, kes saavad madalaid ja keskmisi sagedusi (kuni 1-0,24 Hz);
4. rühm - lapsed, kes tajuvad laia sagedusvahemikku (kuni 2-0,48 Hz ja rohkem)
Seega, kui kuulmise sagedus suureneb, suureneb selgelt võime hääle- ja kõnehelide eristamiseks ning ainult madalate sageduste (rühmad 1 ja 2) juuresolekul kõnehelide eristamise võime praktiliselt puudub.
- Kuulmispuudega laste pedagoogiline klassifikatsioon
Arendaja L. M. Boskis. See põhineb kõne arendamise põhimõttel. Ta tõi kurtide ja vaegkuuljate kategoorias välja kaks laste rühma. Kurtide seas:
- kurt ilma kõneta (kurt ja tumm);
- kurt, kinnipeetud kõne (hiline kurt);
kuulmispuudega inimeste kahjustused:
- kuulmispuudega, arendanud kõnet väikesed vead(grammatika kõrvalekalle, kirjavead ja hääldus);
- kuulmispuue koos sügava kõne alaarenguga (üksikute sõnade kasutamine, vale konstruktsiooniga lühikesed fraasid)
Kõne arengu tase sõltub kuulmislanguse astmest, kuulmisfunktsiooni kahjustamise ajast, tingimustest, milles laps on enne kooli, ja individuaalsetest omadustest.
Boskise tähelepanekute kohaselt võib eristada järgmisi perioode:
- kuulmislangus kuni 1,5-2 aastat, s.t. enne kõne moodustamise perioodi viib kõne täieliku puudumiseni (kõne moodustamise tingimusliku perioodi määrab vanus 2 kuni 7 aastat);
- kuulmislangus 3 kuni 3 aastat, toob kaasa kõne kadumise, mis on juba tekkinud, kui lapse kuulmine vastas normile;
- kuulmislangus 4-5-aastaselt põhjustab peaaegu täieliku kõne kaotuse, kui selle säilitamiseks ei võeta meetmeid;
- kuulmislangus 7. eluaastaks, kui kõne moodustamine on põhimõtteliselt lõppenud, suurendab selle säilimise tõenäosust;
- kuulmislangus pärast 7 aastat, kui lapsed on juba kirjaoskuse omandanud, võivad luua tingimused kõne säilitamiseks koos süstemaatilise tööga.
Tingimused lapse kasvatamiseks enne kooli mõjutavad oluliselt kõne arengut. Mida varem antakse lapsele kõne valdamisel kvalifitseeritud abi, seda edukam on selle kujunemine. Lapsed, kes külastavad eriasutusi: lasteaiad, lasteaiad, rühmad laborites kuulmise ja kõne arendamiseks jne.
Ja ka vanemate sihipärane töö nendega aitab kaasa laste kõne edasisele edukale arengule koolis.
Seega on kõne arengutase üks juhtivaid kriteeriume kuulmispuudega laste omistamisel vastavatesse rühmadesse.
mul on piiratud võimalused kuulmisimitatsiooni puhul näitavad väikelapsed kalduvust visuaalselt-lihaste jäljendamisele, kõne liigutustele, mida nad näevad ka teistel.
Kõigest öeldust järeldub, et kurtusega kaasnevad suulisele kõnele ja selle arengule lapsel nii tõsised tagajärjed (kuigi kõneaparaat ei ole kahjustatud), et ilma eripedagoogilise sekkumiseta osutuvad need ületamatuks.
- Laste kuulmispuude ennetamine
Laste püsiv kuulmispuue on kuulmisorgani varasemate haiguste või pärilike defektide tagajärg. Ravi tegevused enamikul juhtudel osutuvad need ebaefektiivseks. Psühholoogiline, meditsiiniline ja pedagoogiline korrektsioon ja taastusravi, samuti kuuldeaparaadid annavad teatud tulemusi, kuid ei kompenseeri täielikult kuulmiskahjustust. Seetõttu usuvad arstid ja kurtuse õpetajad, et on vaja võtta meetmeid kuulmislangust ja kurtust põhjustavate tegurite ärahoidmiseks ja kõrvaldamiseks.
Seega on kuulmiskahjustusi võimalik vältida ja nende põhjuste kõrvaldamine tooks kaasa kuulmislanguse ja kurtusega laste olulise vähenemise. WHO andmetel oleks pooltel juhtudel olnud võimalik kuulmislangust vältida kõige lihtsamate vahenditega (tabel nr 12-13-14)
Lapse esimesed eluaastad on kõne arengus paljudes aspektides kriitilised, mistõttu kuulmispuue on esmatähtis. Esimese eluaasta laste kuulmispuude õigeaegne avastamine viib kurtide ja tummuse tekkeni ning sellest tulenevalt ka laste puude tekkimiseni.
Vastsündinute patoloogia ja enneaegse sünnituse osakonna sünnitusmaja neonatoloog, lähtudes vähemalt ühe kuulmislanguse ja kurtuse riskiteguri olemasolust vastsündinu vahetuskaardil, märgib ära ähvardava kuulmislanguse ja osutab asjaolule. . Lisaks viib neonatoloog pärast haiglast väljakirjutamist vestluse vanematega, juhendades neid uurima last esimesel eluaastal ning haigeid ja enneaegseid lapsi pärast haiglast väljakirjutamist.
Kui kahtlustatakse kuulmislangust, suunatakse laps kurtuskeskusesse audioloogilisele uuringule.
Sellega seoses on väga soovitav viia esimesel eluaastal läbi kõikide laste käitumiskontrolli.
Reeglina ei registreerita ühepoolse ja nõrga kuulmislangusega lapsi audiologopiidsetesse ruumidesse. Need lapsed on aga ohus ja vajavad süstemaatilist jälgimist.
Seetõttu on hädavajalik kehtestada kahepoolse ja ühepoolse kuulmislangusega, sensineuraalse, sega- ja juhtiva kuulmislangusega, samuti mitte ainult raske kuulmislangusega laste varajane (alates esimestest elukuudest) avastamine, kuulmiskahjustuste rehabilitatsioon. ja kurtus, kuid ka nõrga ja mõõdukaga.
Enam kui 50% kaasasündinud sensorineuraalse kuulmislanguse ja kurtuse juhtudest on geneetiline (pärilik) põhjus. Sel juhul võib kuulmispuue kohe sündides puududa ja areneda hiljem, mõjutada ühte või mõlemat kõrva, varieeruda väikestest kaotustest kurtuseni.
Geneetilised kuulmiskahjustused võivad olla progresseeruvad, kaasasündinud ja ilmneda esmakordselt täiskasvanueas; kuuluma sündroomide hulka ja olema sündroomivaba; autosomaalse retsessiivse ja domineeriva pärandiga, X-seotud.
Igal aastal tuvastatakse üha rohkem mutatsioone, mis põhjustavad sensorineuraalseid kuulmiskahjustusi. Nüüd on teada rohkem kui 100 sellist häiret, mis põhjustavad muutusi valkude struktuuris, mis moodustavad peaaegu kõik sisekõrva elemendid: juukserakud, tugirakud, veresoonte stria, basilaarmembraan, spiraalsõlm, kuulmisnärv.
Enamik geneetiliselt määratud sensineuraalseid kuulmiskahjustusi (HHI) on autosoomne retsessiivne ja mittesündroomne. Enam kui 50% mittesündroomsest CHF-st on seotud konnektiini 26 ja konneksiin 30 valkude struktuuri kõrvalekalletega.
Lapse kuulmiskaotust tuleks kahtlustada, kui lapse areng ei vasta järgmistele punktidele:
Vastsündinud - 3 kuud
- reageerib tugevatele helidele
- ärkab helidest
- vilgub või ajab silmad suureks valju müra peale
3-4 kuud
- rahuneb ema hääle peale
- lõpetab mängimise, kui kuuleb uusi helisid
- otsin uute helide allikat, mida pole silmapiiril
6-9 kuud
- mängides muusikaliste mänguasjadega
- ütleb "ema"
12-15 kuud
- teab oma nime ja sõna "ei"
- kasutab aktiivselt 3-5 sõna sõnastikku
- imiteerib mõningaid helisid
18-24 kuud
- tunneb kehaosi
- kasutab aktiivset sõnastikku 2-sõnaliste fraasidega (vähemalt 20–50 sõna)
- 50% lapse kõnest on võõrastele arusaadav
36 kuuks
- kasutades aktiivset sõnavara, mis koosneb 4 lausest ja 5 sõnast (umbes 500 sõna)
- 80% lapse kõnest on võõrastele arusaadav
- saab aru mõnest tegusõnast
Sündroom, autosoomne dominant
- Waarderburgi sündroom on kõige tavalisem autosomaalne dominantne sündroom. Iseloomustatud järgmised sümptomid: telekant, lai väljaulatuv ninasild, sulanud kulmud, vikerkesta heterokroomia, sensineuraalne kurtus, valge juuksekiht lauba kohal, depigmenteeritud laigud nahal. Mõnel juhul täheldatakse ptoosi, alalõug, suulaelõhe või kõrge suulagi, väikesed luustiku deformatsioonid ja südame defektid.
- Branchio-otorenaalne sündroom (BOR-sündroom)-seda iseloomustavad kõrvalekalded väliskõrva, kaela tsüstide, kuulmiskahjustuste ja neerude arengu kõrvalekallete arengus. Arvatakse, et see sündroom moodustab 2% rasketest kaasasündinud kuulmiskahjustustest. Levimus on umbes 1 elanik 40 000 -st. Kõigil sündroomi tunnustel on erinev väljendusrikkus. See tähendab, et juba kohalolek kliinilised ilmingud ja nende raskusaste võib patsienditi erineda, sealhulgas samas perekonnas.
- 2. tüüpi neurofibromatoos - kahepoolsed kuulmisnärvi neuroomid. Sagedus on 1 50 000 elaniku kohta.
- Stickleri sündroom (David-Stickler, Stickler-Wagner) on pärilike kollagenopaatiate rühm (II ja IX tüüpi kollageen). Haigust iseloomustavad näomuutused, silmakahjustused, kuulmislangus ja liigesehäired. Näo lamestamist võib pidada üheks tüüpiliseks ilminguks. Selle põhjuseks on nina silla moodustavate sügoomaatiliste kaared ja luud vähearenenud. Tüüpilised on muutused vastavalt Robini kompleksi tüübile: makroglossia, mikrognaatia, kõva suulae lõhustamine. Silma kuju muutused põhjustavad tõsist lühinägelikkust. Lisaks on eelsoodumus glaukoomi ja võrkkesta irdumise tekkeks. On väljendunud liigeste hüpermobiilsus, valu, artriit. Tüüpiline on selgroo kõverus (kyphosis, kyphoscoliosis). Lisaks näoskeleti muutused, eriti kõva suulae lõhustamine.
- Achondroplasia on pärilik inimese haigus, mis on tuntud juba antiikajast, mis väljendub enchondral luustumisprotsesside rikkumises normaalse epostaalse ja periosteaalse luustumise taustal, mis viib pikkade luude vähearenenud arengu tõttu kääbusesse; mida iseloomustab kaasasündinud kõrvalekallete olemasolu, eriti seljaaju kanali kaasasündinud stenoos.
- Pageti tõbi on pärilik haigus, mida iseloomustab reieluu ja sääreluu, selgroo ja kolju deformatsioon koos raske hüperostoosiga, luude paksenemine ja kõverus ning kasvajate esinemissageduse suurenemine.
Sündroom, autosoomne retsessiivne
- Usheri sündroom - seda iseloomustab kuulmislangus, progresseeruv nägemiskaotus, vestibulaarne düsfunktsioon ning see on enamikul juhtudel kooliealiste laste kurtuse ja pimeduse põhjus.
- Pendredi sündroom - mida iseloomustab kilpnäärmehormoonide biosünteesi rikkumine ja mis väljendub kaasasündinud hüpotüreoidismis, nodulaarne struuma ja CHN.
- Jervell-Lange-Nielseni sündroom on haruldane pärilik haigus, mis avaldub kaasasündinud kurtuse ja mitmete südamepatoloogiate kujul. See sündroom on üks kliinilised vormid PQ intervalli pikenemise sündroom.
- Refsumi haigus - mida iseloomustab raske progresseeruv kesknärvisüsteem ja retiniit pigmentosa, mis on põhjustatud fütaanhappe ainevahetuse häiretest.
X-seotud sündroomne kuulmiskahjustus
- Alporti sündroom - progresseeruv kesknärvisüsteem, progresseeruv glomerulonefriit ja mitmesugused oftalmoloogilised nähud. Kuulmislangus ilmneb tavaliselt alles 10 -aastaselt.
Artikli ettevalmistamisel kasutati järgmisi allikaid: